



Introduction
The glomerular capillary wall acts as a filtration barrier and exhibits selective permeability. The barrier comprises an inner layer of glomerular endothelial cells (GECs), a glomerular basement membrane (GBM), and an exterior layer of visceral epithelial cells named podocytes. GECs, facing the capillary lumen, are very flat cells with numerous fenestrations that facilitate and regulate the functions of filtration [1]. Podocytes, facing the urinary space, are terminally differentiated cells characterized by their large cell bodies, long major processes, and smaller foot processes. The foot processes derived from adjacent podocytes interdigitate with each other, forming a slit diaphragm to cover the capillaries. Between the GEC and podocyte layers, there is a condensed network of extracellular matrix (ECM) called GBM, which is composed preeminently of two heterotrimeric proteins, type IV collagen and laminin, as well as sulfated proteoglycans [2].
Since the high selectivity of the glomerular filter is achieved by the collaboration between each component of the glomerular filtration barrier (GFB), the podocytes and GECs crosstalk is essential not only for the development and maintenance of an efficient filtration process in physiological conditions but also has a fundamental role in promoting or delaying disease progression. In the glomerular microenvironment, abnormality or health of one cell type can influent the nearby cells by signaling molecules and extracellular vesicles (EVs). Secreted growth factors and signaling peptides, which may have an autocrine effect on the same cell type or a paracrine effect on nearby cells, serve as crosstalk effector molecules by binding to their specific receptors and activating signaling [3]. The ECM is crucial for depositing secreted ligands, the development of concentration gradient, and the presentation of ligands to cell surface receptors [4]. In addition to growth factors and signaling peptides, there is growing evidence implying an important role for EVs in cell communication [5]. This review will focus on recent data concerning the crosstalk between GECs and podocytes in physiological and pathological conditions. In particular, we will focus on the role of vascular endothelial growth factor A (VEGF-A), angiopoietins (ANGPTs), CXCL12/CXCR4/CXCR7, endothelin-1 (ET-1), interleukin-6 (IL-6), and EVs.
Vascular endothelial growth factor A signaling
Vascular endothelial growth factor A signaling in renal diseases
VEGF-A is a key factor for angiogenesis in multiple organ systems, including the formation and maintenance of the microvascular beds of the kidney. In Fig. 1, the role of VEGF on glomerular development is presented.
Podocytes begin to express all VEGF-A isoforms at the S-shaped stage of glomerular formation. During the capillary loop stage, premature podocytes express VEGF-A, encouraging the migration of VEGF receptor 2 (VEGFR-2) positive endothelial cell (EC) precursors in the renal mesenchyme. ECs move into the vascular cleft, proliferate, and differentiate in close proximity to the podocytes that produce VEGF-A [6]. Research in mice has demonstrated that decreased VEGF-A signaling from podocytes causes a loss of ECs’ migration and proliferation, which eliminates the GFBs’ functionality and reduces mice survival [7]. In the mature glomerulus, renal thrombotic microangiopathy (TMA) is caused by postnatal podocyte-specific VEGF-A deletion in mice and VEGF-A inhibition in humans, highlighting the significance of a sufficient level of VEGF-A within the mature kidney for maintaining the normal function of renal microvasculature [8].
Beyond the VEGF-A signaling to the glomerular endothelium, there is also an autocrine pathway involving the soluble form of VEGFR-1 (sFlt1) released by podocytes. Through binding to glycosphingolipids in lipid rafts, sFlt1 initiates an intracellular signaling cascade, facilitating actin reorganization and cell adhesion. Interestingly, severe proteinuria and renal failure are caused by the deletion of sFlt1 from podocytes [9].
Numerous kidney diseases involve VEGF signaling. According to recent animal research and clinical observations, endothelial dysfunction in preeclampsia may be brought on by the placenta’s excessive release of sFlt1 into the mother’s bloodstream. In this contest, VEGF-A might be trapped by sFlt1, leading to the reduction of free VEGF-A in circulation. Rats receiving an adenovirus expressing sFlt1 developed proteinuria, glomerular endotheliosis, and hypertension [10]. Podocyte-specific VEGF-A haploinsufficiency in mice causes proteinuria, endotheliosis, and, finally, the loss of ECs, similar to the characteristic renal lesions found in preeclampsia [11]. In humans, sFlt1 levels begin to rise at least 5 weeks before the onset of preeclampsia and remain elevated [12]. The finding that therapy with neutralizing VEGF-A antibodies can be associated with glomerular endothelial damage, endotheliosis, and proteinuria further supports this correlation [13].
The kidney and brain are especially affected by TMAs, a group of related illnesses in which the development of intracapillary and intra-arteriolar platelet thrombi results in end-organ ischemia and infarction. Hemolytic uremic syndrome (HUS), a kind of TMAs, is characterized by the formation of fibrin-platelet thrombi and damage to the ECs, including ballooning, detachment, and endotheliosis. It is essential to highlight that individuals taking anti-VEGF drugs for cancer may experience kidney histology abnormalities that resemble TMAs [13].
Furthermore, it appears that VEGF has a role in developing diabetic nephropathy (DN). The glomerulus displays higher VEGF-A levels in the early angiogenic stage of DN. Experimental models of early diabetes have revealed glomerular overexpression of VEGF-A and its receptors [14], and markers of DN can be attenuated by blocking VEGF-A in rodents [15]. Moreover, transgenic overexpression of VEGF-A in podocytes causes the GBM to be thickened, proteinuria, and DN hallmarks [16]. As mentioned before, the sFlt1 acts as an antagonist of VEGF-A through sequestering circulating VEGF-A. In experimental diabetes, inducible overexpression of sFlt1 in podocytes of mice results in a reduction of albuminuria and amelioration of glomerular alterations [17]. In contrast to these findings, it has also been observed that the specific deletion of podocyte-VEGF-A accelerates renal damage in an experimental model of diabetes [18] and a decrease of VEGF-A expression in human diabetes [19]. These findings revealed that, depending on the signal intensity, there might be a delicate balance between the protective and harmful effects of VEGF-A.
VEGF signaling is probably involved also in crescentic glomerulonephritis (CGN) and membranoproliferative glomerulonephritis (MPGN). High serum and urine levels of VEGF are reported in patients with CGN [20]. In human MPGN, VEGFR-1, VEGFR-2, and neuropilin-1 are expressed in mesangial cells (MCs), and VEGF-A can induce MC proliferation [21].
Vascular endothelial growth factor A signaling in kidney transplantation
Particularly interesting is the potential role of VEGF in kidney transplantation. Earlier investigations showed that human chronic allograft nephropathy (CAN) and experimental models both exhibit elevated expression of VEGF in the interstitial cell [22]. According to Malmström et al. [23], the chronic allograft damage index (CADI) score was correlated with the total intragraft as well as interstitial inflammatory cell expressions of VEGF and VEGFR-1. PTK787’s inhibition of the VEGF receptor significantly reduced both the CADI score and fibrosis. This finding suggested that elevated VEGF activity may facilitate alloimmune-induced inflammatory responses, ultimately resulting in fibrotic changes [23]. Moreover, VEGF may accelerate allograft vasculopathy by enhancing smooth muscle cell (SMC) migration either directly or by increasing the production of platelet-derived growth factor (PDGF) by surrounding cells [24]. In addition to its direct atherogenic effects, VEGF is crucial in controlling the mobilization, homing, and differentiation of vascular progenitor cells. Circulating VEGFR-2 positive progenitor cells transform into ECs or SMC when stimulated with VEGF or PDGF-BB, respectively, which could hasten the onset of allograft atherosclerosis [25]. It seems that the effects of VEGF on renal allografts are time-dependent. A study from Ozdemir et al. [26] demonstrated that in the short term after kidney transplantation, both tubular and interstitial VEGF expression acts as a protective signal on renal allografts. However, over the long term, interstitial fibrosis (IF) and, consequently, poor graft outcomes would be more likely in patients with marked tubular and interstitial VEGF expression [26].
Endothelin-1 signaling
Endothelin-1 signaling in renal diseases
The human kidney expresses all three ET family members, ET-1, ET-2, and ET-3, albeit ET-1 is the most common isoform [27]. ETA receptors (ETAR) and ETB receptors (ETBR) are two G-protein-coupled receptors that ET-1, ET-2, and ET-3 bind to, whereas ET-3 has a low affinity for the ETAR at physiological concentrations [28]. In the vasculature, ETAR predominantly mediates vasoconstriction and mitogenesis, whereas ETBR mainly mediates vasodilation and inhibition of growth and inflammation.
The human kidney expresses a high density of ETBR [29]. By engaging in ETBR, ET-1 releases vasodilators in an autocrine or paracrine manner. Moreover, the kidney, liver, and lung endothelial ETBR play a critical role in scavenging ET-1 from the plasma [30]. The activation of ETBR in medullary epithelial cells, which lowers salt and water reabsorption, is the third important role of ET-1 [31]. ETAR and ETBR are also present in human and rat podocytes and MCs [32]. ET signaling in podocytes is involved in different renal diseases, such as DN [33], proliferative lupus nephritis (LN) [34], and focal segmental glomerular sclerosis (FSGS) [35]. Lenoir et al. [33] showed that in mice with podocyte-specific double deletion of the alleles of ETAR and ETBR, diabetes-induced glomerulosclerosis and podocyte loss are avoided. Additionally, they discovered that ET-1 could directly activate the nuclear factor kappa B and β-catenin pathways in podocytes, which promotes the development of diabetic glomerulosclerosis and the loss of podocytes [33].
Results from the histological examination of LN samples pointed to a correlation between the width of the foot process and the pathological score of GEC damage. More ET-1 was secreted when GECs were exposed to a podocyte-conditioned medium stimulated with immunoglobulin G (IgG) from LN patients (PCM-LN). A redistribution of cytoskeleton F-actin and a marked decrease in nephrin was noted when podocytes were exposed to an endothelial-conditioned medium stimulated with PCM-LN (ECM-PCM-LN). It should be emphasized that the anti-ETAR antibody could block these effects, demonstrating that GECs and podocytes communicate among themselves through ET signaling [34].
Moreover, ET-1 plays a role in FSGS, frequently accompanied by proteinuria and a steady decline in glomerular function. Podocyte damage, podocyte depletion, and glomerular capillary segment collapse are symptoms of FSGS. Studies performed by Daehn et al. [35] and Ebefors et al. [36] demonstrated that in transgenic mice and BALB/c mice with adriamycin-induced glomerulosclerosis, podocyte-specific activation of transforming growth factor-beta (TGF-β) can cause ET-1 release by podocytes and enhance ETAR expression in nearby ECs. The paracrine ETAR activation by ET-1 led to degradation of the glomerular endothelial surface layer, mitochondrial oxidative stress, and dysfunction of GECs. ETAR antagonism prevented all of these consequences. Albuminuria, glomerulosclerosis, and podocyte apoptosis were, in turn, promoted by endothelial dysfunction [35,36].
Endothelin-1 signaling in kidney transplantation
Ischemia-reperfusion injury (IRI) and acute and chronic rejection after kidney transplantation can all trigger the innate and adaptive immune response. ET-l synthesis in vitro is impacted by a variety of cytokines that are released by infiltrating activated mononuclear cells. Tumor necrosis factor-alpha (TNF-α) can increase the ET-1 messenger RNA (mRNA) and ET-l protein release in rat MCs [37]. In bovine ECs, interferon-gamma cotreatment potentiates the TNF-α effect by boosting ET-1 synthesis [38]. TGF-β can stimulate ET-l secretion in cultured MCs [39], endothelial [40], glomerular epithelial [41], as well as tubular epithelial cells (TECs) [42]. These results suggest that different cytokines and growth factors can regulate ET expression in the allograft during the immunological response to alloantigen stimuli. Also, it was discovered that upregulation of ET-1 and its receptors in experimental and human kidney transplantation. Recent research has shown that chronic renal allograft rejection in rats results in a considerable overexpression of ET-1, ET-3, and their receptors. Moreover, in these renal allografts, inhibiting ETs reduced chronic rejection, suggesting a potential role for ETs in the pathogenesis of CAN [43]. CAN is associated with a higher level of ET-1 in human kidney transplantation [44]. Medial SMCs and SMCs within the neointima both displayed elevated expression of ETAR in intrarenal arteries with transplant renal arteriosclerosis. These results suggest that increased ETAR expression may enhance the local proliferative and vasoconstrictive effects of ET-1 in human kidney allografts [45].
Paracrine signaling between vascular endothelial growth factor A, endothelial nitric oxide synthase/nitric oxide, and endothelin-1
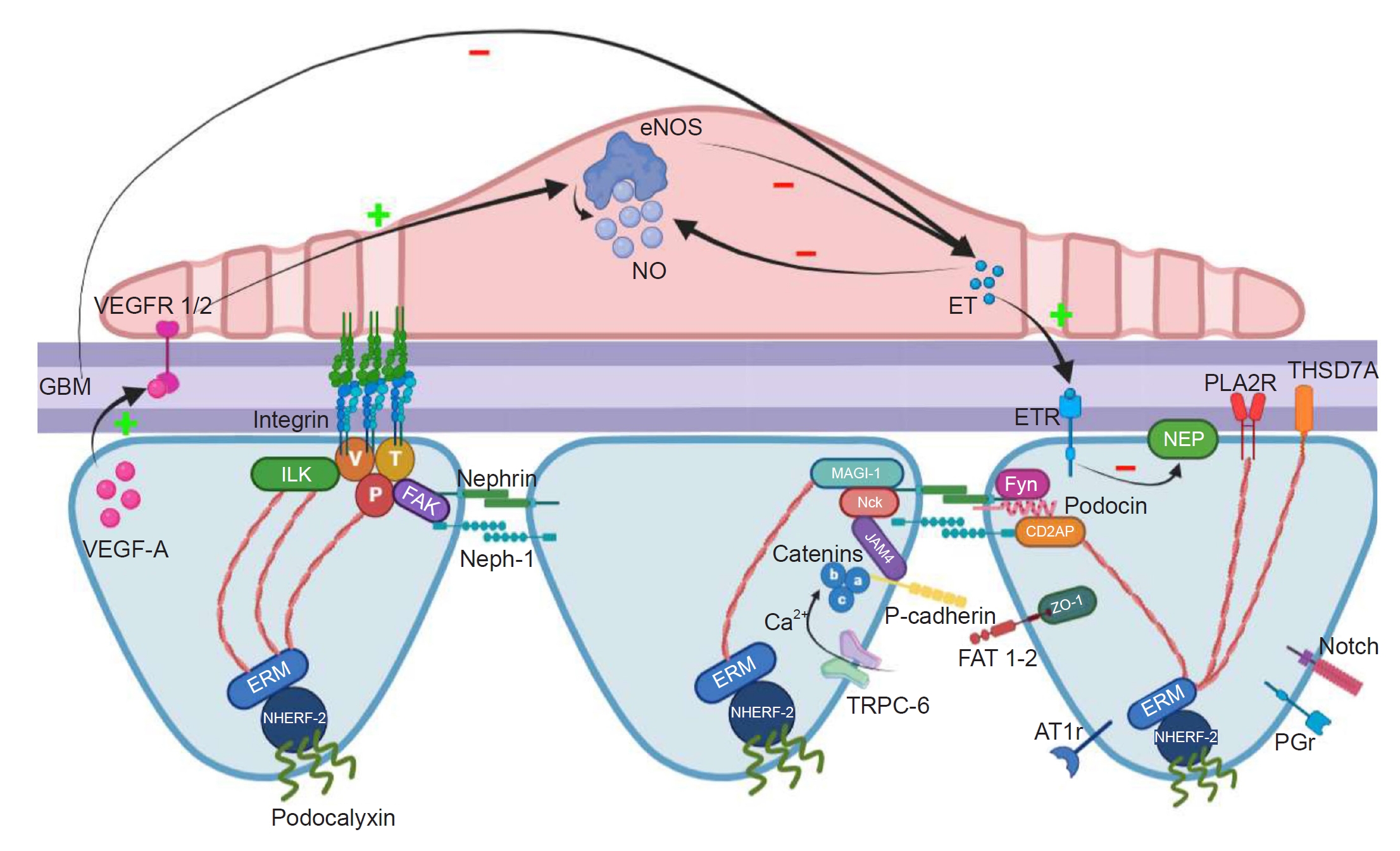
It has been shown that nitric oxide (NO) has antithrombogenic properties, inhibiting EC activation/injury brought on by cytokines and promoting vasodilatation, all of which are protective for the vascular system. The main sites of endothelial NO synthase (eNOS) expression in rodents and humans include the medullary vasa recta, glomerular and peritubular capillaries, afferent/efferent arterioles, and the endothelium of intrarenal arteries [46,47]. Mice lacking eNOS displayed podocyte damage and aberrant mitochondria [48]. The protection of podocytes from TNF-α induced loss of synaptopodin by conditioned medium from eNOS-overexpressing microvascular ECs in vitro suggests that healthy GECs protect podocytes from inflammatory insults in a paracrine manner by secreting protective mediators [49]. Furthermore, the preservation of glomerular integrity may rely on the paracrine signaling between VEGF-A, eNOS/NO, and ET-1 in podocytes and GECs. Under physiological conditions, through binding to its receptors VEGFR-1 and VEGFR-2 expressed on GECs, VEGF-A synthesized by podocytes can induce eNOS activation in GECs and subsequently increase NO production [8]. The increase of NO may negatively regulate the amount of VEGF-A produced by podocytes [50]. The glomerular cells control the proper VEGF-A production through this crosstalk, preventing excessive vascular growth while maintaining VEGF-A availability.
In addition to NO, VEGF-A also regulates ET-1 production by GECs. A study by Collino et al. [51] revealed that podocyte VEGF-A blockade causes ET-1 release from GECs. High levels of ET-1 prevent the formation of NO, while low levels of ET-1 promote its production [52]. In addition to cytoskeleton rearrangement, ET-1 produced from GECs can also result in a decrease of nephrin in podocytes [34]. Conversely, NO has protective effects on podocytes and lowers the expression of ET-1 [49]. An illustration of crosstalk between GECs and podocytes in the VEGF-A-eNOS/NO-ET-1 axis is shown in Fig. 2.
Angiopoietin signaling
Angiopoietins in renal diseases
ANGPTs belong to the vascular growth factor family, including ANGPT1, ANGPT2, ANGPT3, and ANGPT4. ANGPT1 works as a Tie2 receptor agonist, supporting an anti-inflammatory, pro-survival, and anti-permeability phenotype of the vasculature. Contrarily, ANGPT2, secreted by ECs in response to proinflammatory stimuli, prevents Tie2 from being phosphorylated and thus breaks up the protective Tie2 signaling. Hence, it appears that the equilibrium between ANGPT1 and ANGPT2 is what controls signaling through Tie2.
Much like the VEGF-VEGFR paracrine pathway, ANGPT1 is expressed by podocytes and MCs, while its target Tie2 is expressed by GECs. ANGPT1 is essential for maintaining healthy glomeruli, and signaling of the ANGPT1/Tie2 pathway appears essential for maintaining the filtration barrier both during the normal development of the kidneys and during pathological circumstances.
Global deletion of ANGPT1 before the embryonic day (E) 12.5 causes vascular abnormalities that lead to early embryonic death. The glomerulus of mice with induced ANGPT1 deletion at E10.5 showed abnormalities, including dilated capillary loops, an unorganized GBM structure, and MC reductions, while podocytes appeared intact. The global ANGPT1 deletion induced later did not result in any obvious phenotype [53]. This indicates that ANGPT1/Tie2 signaling is required for the development of the vasculature, including glomerular capillaries, but not necessary for quiescent vessels. In pathological conditions like diabetes, ANGPT1-deficient diabetic mice displayed higher proteinuria, mesangial matrix expansion, and glomerulosclerosis compared to diabetic controls. Albuminuria and GEC proliferation were delayed by the repletion of the glomerular ANGPT1 in diabetic mice with selective podocyte-specific overexpression of ANGPT1 [54].
Moreover, a decrease in the endothelial survival markers VEGF-A and ANGPT1 and an increase in ANGPT2 were temporally associated with the loss of glomerular capillaries in a mouse model anti-GBM glomerulonephritis [55]. These findings imply that ANGPT1/Tie2 signaling is not only crucial for maintaining GFB function but also has a remarkable capacity to regulate the glomerular capillary response to damage. Contrarily, the overexpression of ANGPT2 in podocytes increases GEC apoptosis and albuminuria, indicating that ANGPT2 may compete with ANGPT1 [56].
Angiopoietins in kidney transplantation
Increased ANGPT2 levels (the natural Tie2 antagonist) have been demonstrated to correlate with mortality in kidney transplant recipients, suggesting that an unbalanced ANGPT/Tie2 system may be detrimental to renal transplantation [57]. Ma et al. [58] showed that ANGPT1 was downregulated in a rat model of CAN, whereas ANGPT2 and Tie2 were increased. These changes have a strong correlation with the Banff score. Exogenous delivery of a PEGylated synthetic Tie2 agonistic peptide can enhance graft function in a mouse major histocompatibility complex-mismatched renal transplant model by controlling endothelial activation and the transmigration of deleterious inflammatory cells into the interstitium of the transplant [59].
CXCL12/CXCR4/CXCR7 signaling
CXCL12/CXCR4/CXCR7 signaling in renal diseases
Homeostatic chemokine CXC chemokine ligand 12 (CXCL12; stromal cell-derived factor 1) signals through its receptors CXCR4 and CXCR7 [60]. After being stimulated by their same ligand, CXCL12, both receptors can activate multiple cell signaling pathways and/or scavenge CXCL12 from the extracellular space. This promotes the development of organs and the preservation of homeostasis. In the kidney, podocytes can produce CXCL12, which acts on CXCR4 expressed by GECs and carries out an essential role in podocyte-EC cross-communication. Nephrogenesis, particularly in the formation of the renal vasculature, strongly benefits from CXCL12/CXCR4/CXCR7 signaling [61]. Similar renal phenotypes with abnormal blood vessel development, like the ballooning of glomerular capillaries and altered conformation of the renal vasculature, were seen in mice lacking either CXCL12 or CXCR4. Deletion of CXCR7 in mice, which also leads to a reduction of CXCR4 expression, replicated the phenotype of the CXCR4 deficient mice, suggesting that CXCR7 closely controls CXCL12/CXCR4 mediated signaling between podocytes and glomerular capillaries [62]. Moreover, CXCL12 has a role in several kidney diseases, including renal cell carcinoma, DN, LN, diarrhea-associated HUS, and acute kidney injury (AKI) [63].
CXCL12/CXCR4/CXCR7 signaling in kidney transplantation
According to Hoffmann et al. [64], compared to healthy transplant kidneys, the expression of CXCL12 was considerably higher in transplants with persistent fibrotic lesions. Indeed, CXCL12/CXCR4 induces renal TECs-mesenchymal transition (EMT) with the involvement of the Wnt pathway [65]. Renal allograft fibrosis can be successfully mitigated using a CXCR4 antagonist or a neutralizing antibody [66]. In IRI-induced renal transplantation damages, anti-CXCL12 antibodies could reduce IRI and chronic rejection [67]. On the other hand, studies also revealed that CXCL12 is essential for CXCR4-positive cells, such as hematopoietic stem cells and a fraction of mesenchymal stem cells (MSCs), to home and migrate to the kidney, representing a potential prevention for IRI-induced acute/chronic rejection and maintaining renal function [68].
Interleukin-6 signaling pathway
Interleukin-6 signaling pathway in renal diseases
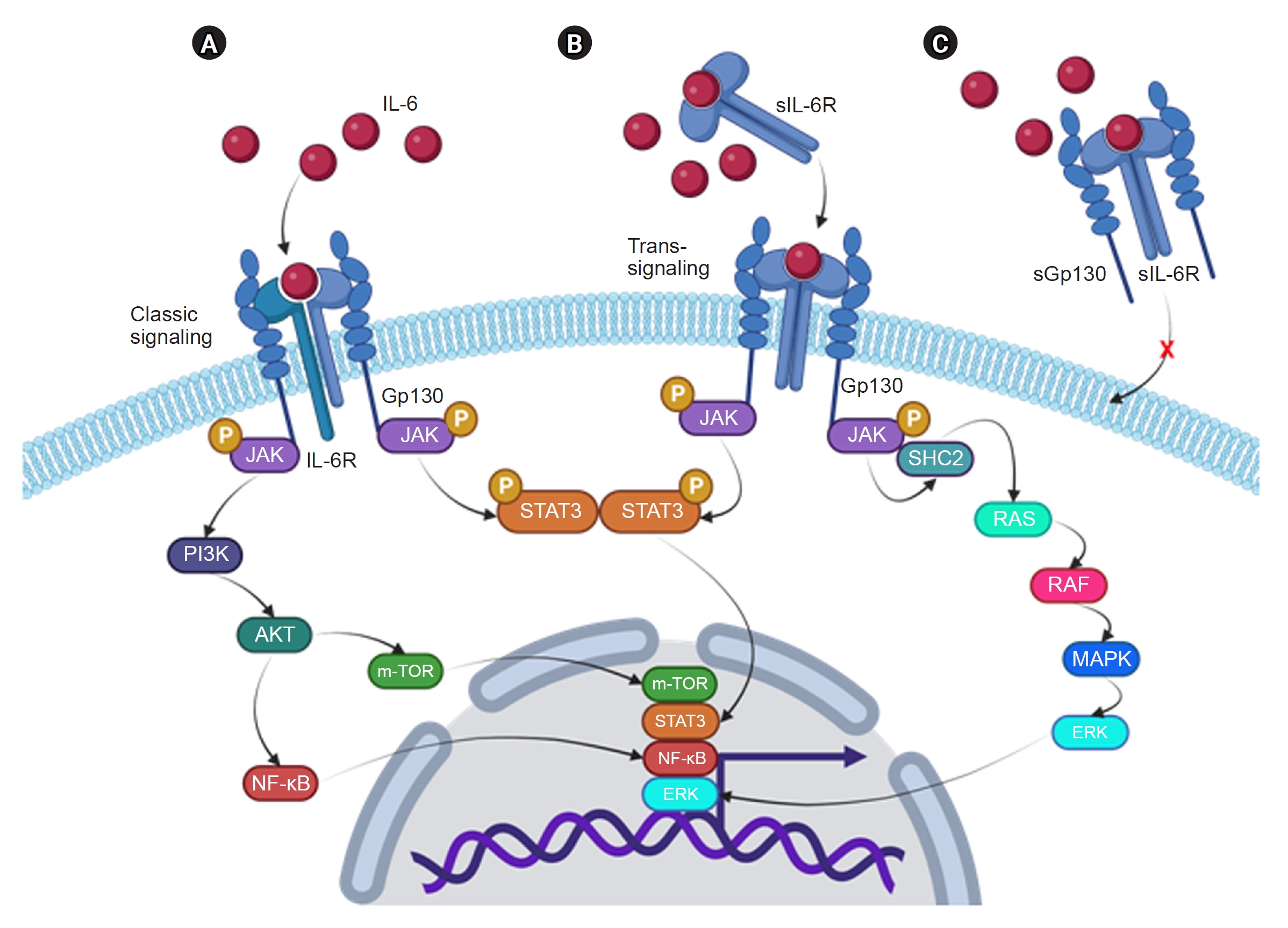
IL-6 is a pleiotropic cytokine that, in addition to immunological and inflammatory responses, also controls hematopoiesis, metabolism, and organ development. Fig. 3 summarizes the two signaling pathways of IL-6: the classical and trans-signaling pathways. The classical pathway is activated by the binding of IL-6 with the membrane-bound IL-6R (mIL-6R, also named CD126 or gp80), while the trans-signaling is induced by the interaction of IL-6 with a soluble form of IL6R (sIL-6R). It’s important to note that there is a naturally produced isoform of Gp130 called soluble Gp130 (sGp130), which is identified in the bloodstream at relatively high concentrations (100–400 ng/mL in human plasma) [69]. sGp130 functions as a specific inhibitor of the IL-6 trans-signaling pathway because it can interact with the IL-6/sIL-6R [70]. It is generally accepted that IL-6 classical signaling is anti-inflammatory and trans-signaling is proinflammatory [71], although there is a debate [72].
Renal resident cells, such as podocytes, ECs, MCs, and TECs, can release IL-6 under certain conditions. Since only podocytes express the mIL-6R, which indicate that only podocytes can respond to IL-6 via both classical and trans-signaling pathway, while in ECs, MCs, and TECs, the IL-6 trans-signaling pathway is predominant. IL-6 signaling is involved in many kidney diseases, such as IgA nephropathy, LN, DN, AKI, and chronic kidney disease [73].
Usually, different pathological stimuli can induce renal resident cells secreting IL-6, which in turn triggers the growth of MCs, the recruitment of inflammatory cells, and the overexpression of the angiotensin (Ang) II type 1 receptor in ECs with the consequence of Ang II-induced vasoconstriction, reactive oxygen species production, and endothelial dysfunction [74].
Interleukin-6 signaling pathway in kidney transplantation
It is widely known that IL-6 has a role in both acute and chronic kidney allograft rejection. Renal expression of IL-6 was elevated with decreased intragraft Foxp3+ Tregs after allograft rejection in a mouse kidney transplant model [75]. Furthermore, the absence of donor-produced IL-6 increased the survival of the renal allograft. It was correlated with higher levels of intragraft Tregs and lower levels of circulating anti-graft alloantibodies, indicating that selective inhibition of donor IL-6 signaling may prevent both humoral and cellular rejection [76]. In an experimental model of CAN, IF and tubular atrophy were demonstrated to be mediated by intragraft B-cell production of chemokines and cytokines, including IL-6. This finding indicates that IL-6 may play a potential role in causing CAN [77]. Renal allograft rejection in human kidney transplant patients is accompanied by increased IL-6 levels in the blood, urine, and biopsy tissue [78,79]. Rejection occurred in renal allograft recipients who developed high blood IL-6 and IL-17 levels during tolerance induction utilizing a mixed chimerism method, but recipients without high IL-6/IL-17 experienced long-term survival without rejection [80]. What’s more noteworthy is that donor genotypes for IL6 and IL6R, but not recipient genotypes, serve as an independent predictive biomarker for biopsy-confirmed renal allograft rejection [81].
It’s important to note that IL-6 signaling seems to have a protective role by boosting the repair process in some pathological circumstances, such as the ischemia-reperfusion–induced AKI model. By an underlying anti-oxidative stress mechanism, stimulation of IL-6 trans-signaling dramatically lowers kidney damage and protects renal function [82]. More interestingly, Kuravi et al. [83] proposed the existence of a crosstalk between podocytes and ECs via IL-6, which was demonstrated in a podocyte-EC coculture system. Particularly, TNF-α stimulated podocytes to release IL-6, which increased the expression of suppressor of cytokine signaling 3 in glomerular endothelium and induced IL-6’s immunosuppressive effect, hence limiting the migration of neutrophils to the endothelium [83].
Extracellular vesicles
Extracellular vesicles: a novel frontier in renal diseases
Recently, cell-cell communication mediated by EVs is an emerging biological concept. Almost all cells secrete EVs, which are divided into exosomes and microparticles based on their size. Contrary to signaling molecules secreted by the cells, which are well-defined proteins with specific roles, EVs contain a concentrated complex of molecules, including lipids, nucleic acids, proteins, glycans, and metabolites. These molecules can exert contemporarily synergistic or antagonistic functions. The EVs cargo is protected from enzymatic degradation in the extracellular environment and can be delivered to distant cells. Moreover, EVs-mRNA can be horizontally transferred to the target cells and translated into the corresponding protein [84]. Therefore, cell-to-cell communication, including podocytes and GECs bidirectional crosstalk through EVs, is an intriguing research topic. Wu et al. [85] found that high glucose (HG) causes GECs to undergo the endothelial-to-mesenchymal transition (EndMT), and HG-treated cells with the EndMT produce more exosomes than normal glucose-treated GECs. They demonstrated that exosomes originating from GECs undergoing EndMT might be taken up by podocytes and can cause the podocyte to undergo epithelial-to-mesenchymal transition (EMT) and barrier failure. Moreover, their study revealed that TGF-β1 mRNA is more abundant in exosomes from HG-treated GECs and likely causes EMT and malfunctioning of podocytes via canonical Wnt/β-catenin signaling. Their findings imply that renal fibrosis in DN is contributed by the paracrine communication between cells undergoing the EndMT and podocytes via exosomes. Therefore, protecting GECs from the EndMT and inhibiting TGF-β1-containing exosome release from GECs could be a new therapeutic strategy to prevent renal fibrosis in DN [85]. According to recent research by Medica et al. [86], an L-selectin-based mechanism was primarily responsible for the internalization of EVs produced from endothelial progenitor cells (EPCs) in both GECs and podocytes. By modifying gene expression and triggering the release of growth factors like VEGF-A and hepatocyte growth factor, EVs improved the development of capillary-like structures and cell migration in GECs. EPC-derived EVs defended GECs against apoptosis in the presence of cytokines such as IL-6, TNF-α, and complement protein C5a by reducing oxidative stress and blocking leukocyte adherence by limiting the production of adhesion molecules (ICAM-1, VCAM-1, E-selectin). In podocytes, EVs reduced apoptosis and blocked the loss of nephrin brought on by cytokines and C5a. More intriguingly, EPC-derived EVs protected podocytes from apoptosis and a change in perm selectivity linked to inflammation-mediated damage in a coculture system of GECs/podocytes that simulated GFB. Moreover, pretreating EVs with RNase rendered their protective actions ineffective, indicating the critical role of RNA transfer from EVs to injured glomerular cells. Their findings suggested that the EPC-derived EVs protected GFB integrity against complement- and cytokine-induced damage, indicating a potential role as therapeutic agents for drug-resistant glomerulonephritis [86].
Extracellular vesicles in kidney transplantation
Recently, the relevance and role of EVs in renal transplantation have attracted increasing attention. Finding the diagnostic and prognostic biomarkers for evaluating donor kidney quality, graft function, and kidney allograft rejection were the main goals of the EV investigations. Turco et al. [87] demonstrated that specific populations of EVs derived from renal parenchymal cells identify kidney structural changes (nephron hypertrophy and nephrosclerosis) through profiling urinary EVs in 138 kidney donors at the time of live-donor transplantation, which may allow clinicians to assess donor kidney health and predict graft function. Using proteome and micro RNA (miRNA) analysis, Lozano-Ramos et al. [88] found that urinary EVs from live donors had higher concentrations of the miR-326 (which targets the B-cell lymphoma 2-related apoptotic pathway) than those from deceased donors. It’s interesting to note that EVs can also be found in donors’ preservation fluid both after circulatory death and after brain death. In the first 7 days after transplantation, miRNA profiling in EVs may be related to graft function [89]. In a recent study, Franzin et al. [90] analyzed the miRNA expression profile of EVs extracted from the plasma of patients with antibody-mediated allograft rejection (AMR). Their research has shown that AMR-derived EVs can cause EndMT and tubular senescence. According to the miRNA expression profile of EVs, miR-604, miR-515-3p, miR-let-7d-5p, and miR-590-3p were upregulated, while miR-24-3p and miR-29a-3p were downregulated. Tubular senescence and EndMT were entirely reversed by the RNase-mediated digestion of EV cargo, indicating that EVs play a significant role in the inflammatory, aging, and profibrotic kidney response during the development of AMR via their miRNA content [90].
Research on EVs has also concentrated on their capacity to provide early prognostic information regarding the outcomes of kidney transplants, such as in situations of chronic renal allograft dysfunction or delayed graft function. The EVs derived from a variety of sources, including MSCs [91], human-induced pluripotent stem cells [92], renal tubular cells [93], and EPCs [94], were shown to reduce IRI through antiapoptotic, anti-oxidative, and anti-inflammatory effects, as well as by inducing regenerative programs that lead to renoprotection in IRI.
Glutamatergic signaling
Given the intricate structure of podocytes and the continual stimulation and stress brought on by blood pressure and contents, these cells probably need a precise and quick modality of communication among themselves and with the other glomerular cells as well. Studies from our group have demonstrated that podocytes possess glutamate-containing vesicular structures that undergo spontaneous and regulated exo-endocytosis [95]. Deletion of Rab3A, a small GTPase that controls glutamate exocytosis, or blocking the glutamate ionotropic N-methyl-D-aspartate receptor (NMDAR) with specific antagonists can alter glutamatergic signaling in podocytes, which can cause significant cytoskeletal reorganization, nephrin redistribution, and an elevated urinary albumin/creatinine ratio in mice [96]. These findings imply that the GFB’s integrity is sustained by glutamatergic signaling mediated by podocytes. Moreover, using a mouse podocyte-EC coculture system, which mimics the GFB in vitro, we demonstrated the existence of a crosstalk between those two cell types via glutamate signaling. We confirmed that the ionotropic glutamate receptor NMDAR and the metabotropic receptor Grm1 are present in mouse GECs in vivo as well as the ECs in culture. The addition of glutamate to the endothelial side of the coculture potently increased albumin permeability and the same effect was obtained by adding to the podocyte medium alpha-latrotoxin, a substance known to induce glutamate release from podocytes. In addition, both treatments caused increased endothelial p44/42 mitogen-activated protein kinase (MAPK). Preincubation of ECs with the NMDAR antagonist MK-801 was able to prevent albumin leakage from the GFB model and abolish 44/42-MAPK activation induced both by glutamate and by alpha-latrotoxin [97]. Those results suggested that excessive activation of EC glutamate signaling can result in the alteration of GFB permeability.
Conclusion
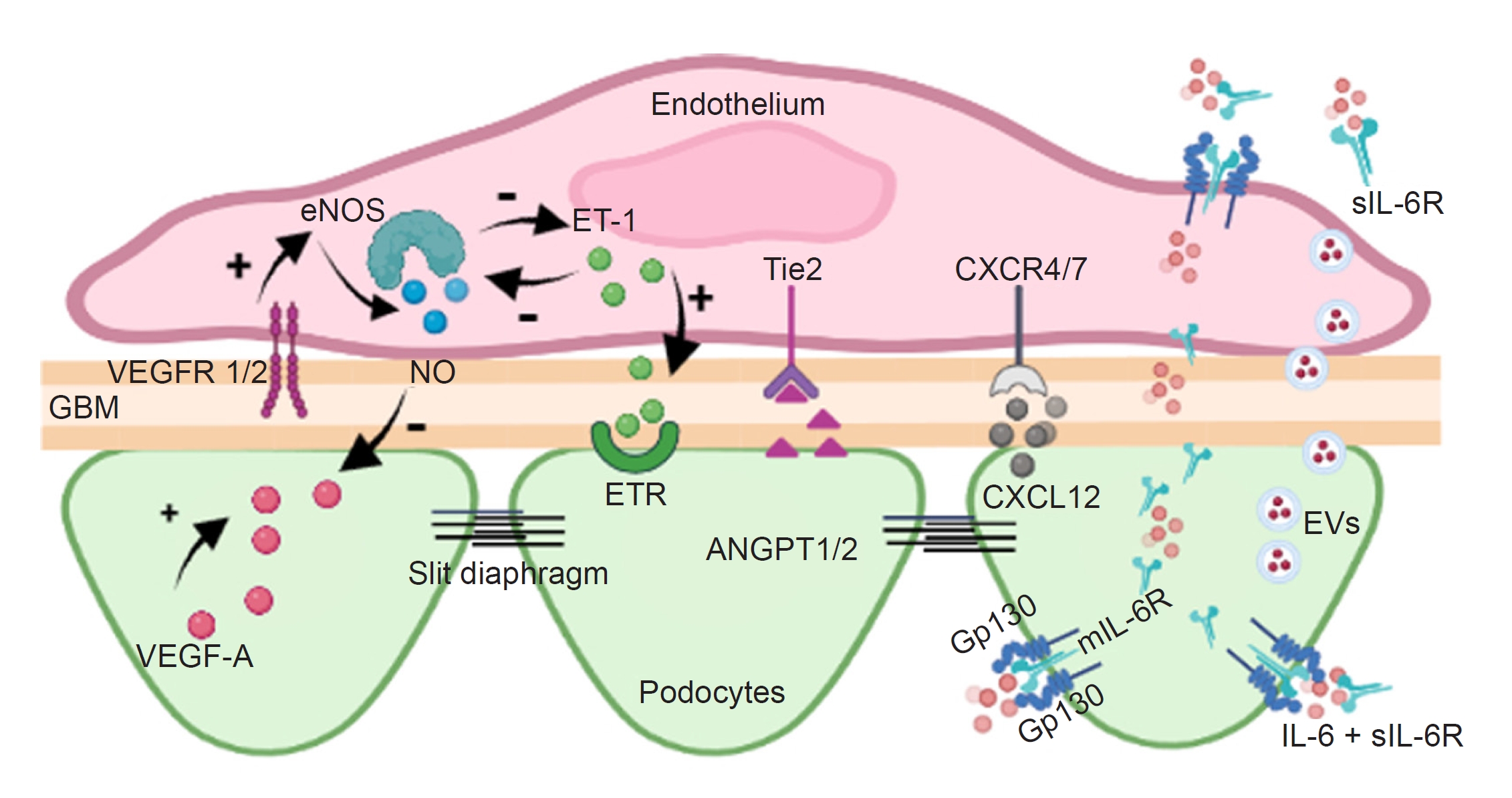
Much evidence suggests that the cell-cell communication between resident cells in the glomerulus through signaling molecules is involved in glomerular homeostasis, renal diseases, and kidney transplants (Fig. 4; Supplementary Table 1 and 2, available online). Our understanding of glomerular cross-communication has substantially expanded with the increased use of cell-specific transgenic models and new techniques, such as genomics, transcriptomics, proteomics, and metabolomics. However, much research must be conducted on discovering novel signaling cascades, particularly those released by EVs from various cell types. The identification of these mediators as well as a better understanding of already established crosstalk molecules, may lead to the recognition of new targets for managing kidney transplantation and the prevention and treatment of glomerular disorders.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement table 1
Supplement table 1 Print
Print






")









