



Overview of renal transplant rejection
Renal transplantation is an effective treatment option for patients with end-stage renal disease. Since the first successful renal transplant, which was carried out between identical twins in 1954 in the United States [1], this procedure has been widely performed in many Western and Eastern countries. In Korea, the first successful renal transplant occurred in 1969, and in the past six years, more than 15,000 renal transplants have been performed [2].
The concept of rejection was recognized in the early 1900s by Alexis Carrell, who coined the term ‘biologic incompatibility’ [1]. Rejection is an immunologic reaction to donor antigens that are recognized by a recipient’s immune system. This continues to present a major obstacle to long-term allograft survival [3], despite progressive improvements in surgical techniques, advances in immunology, and the introduction of new immunosuppressive drugs. Renal allograft biopsy is a direct and confirmative tool for the diagnosis of rejection that can be used to assess both the type and degree of rejection.
Pathologic changes associated with acute and chronic renal allograft rejections have been reported since the late 1960s [4-7]. However, no standardized classification system was proposed until 1991, when a group of 28 renal pathologists, nephrologists, and transplant surgeons gathered in Banff, Canada, and outlined international standards for the definition and grading of transplant rejection [8]. After the first ‘Banff Working Classification of Kidney Transplant Pathology’ report, successive biannual meetings have been held, with the recent 2019 meeting taking place in Pittsburgh [9-18], and the results have been published as meeting reports. In this review, I will discuss morphologic features of renal transplant rejections, up-to-date revisions and pitfalls of the Banff Classification of Allograft Pathology [8-18], and future perspectives.
Pathology of renal allograft rejection
Rejection pathology can be observed in all four components of the kidney—the glomeruli, tubules, interstitium, and vessels—either individually or in combination. In most rejection cases, renal allograft biopsy shows morphologic injuries resulting from predominantly cellular or antibody-mediated mechanisms. Depending on the time post-transplantation and rejection activity, this condition can be classified as either acute/active or chronic. However, acute and chronic changes can exist within the same biopsy [19]. Importantly, although acute and chronic are clinical terms suggesting the time of onset of a disease, here, this classification reflects activity or inactivity, rather than time of biopsy. Thus, active lesions may appear late, and chronic lesions may develop early during the post-transplant period.
Interpretation of allograft histology should consider recipient as well as donor factors. For example, older donors may have significant glomerulosclerosis and tubulointerstitial fibrosis, and kidneys from brain-dead donors may have ischemic changes. Rarely, donor-transmitted disease may be observed in time-zero or early biopsies. In this case, an implantation or zero-hour biopsy may be useful as a reference. In late-transplant biopsies, the possibility of recurrent or de novo development of glomerular, metabolic, or systemic diseases should be considered. Drug toxicity and infections may also develop at any time post-transplant. If a graft biopsy is obtained late, graft histology is likely to show mixed features that are attributable to more than one cause.
Renal allograft biopsy samples can be analyzed by light, immunofluorescence, and electron microscopy. Among these, the features that are assessed by light microscopy are essential and the most important. Immunofluorescence microscopy or immunohistochemistry is needed to detect footprints of antibody binding and immune complexes, whereas electron microscopy is used for the detection of chronic antibody-mediated rejection (ABMR). Rejection pathology can be described according to activity or the histologic component involved, as described below.
Rejection pathology according to activity
Rejection pathology according to histologic component
Glomerulus
Glomerulitis
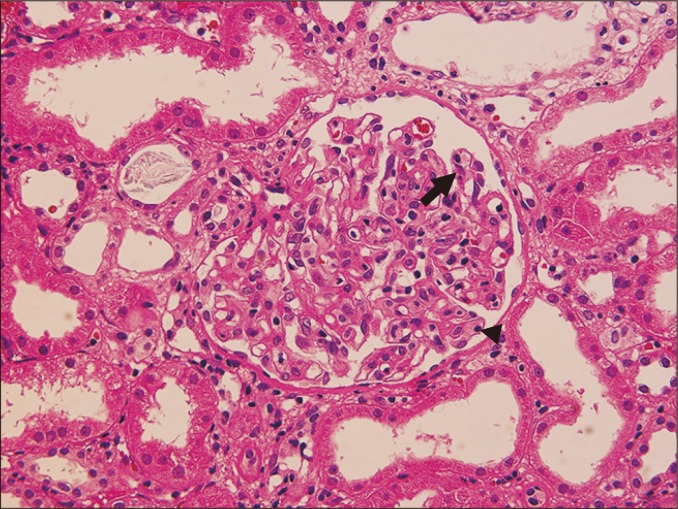
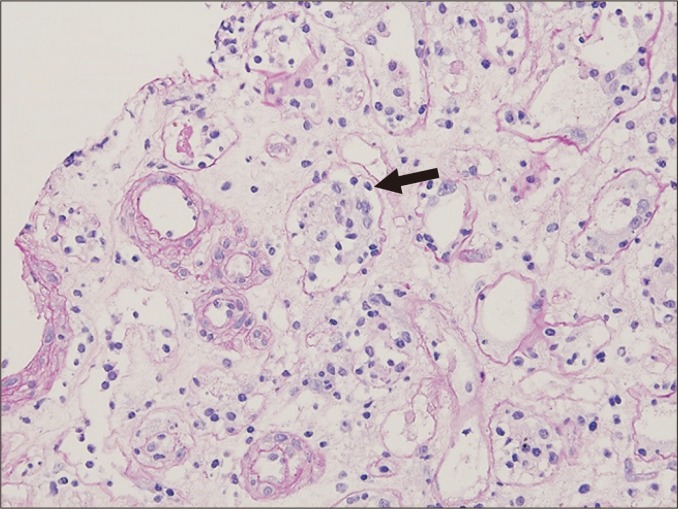
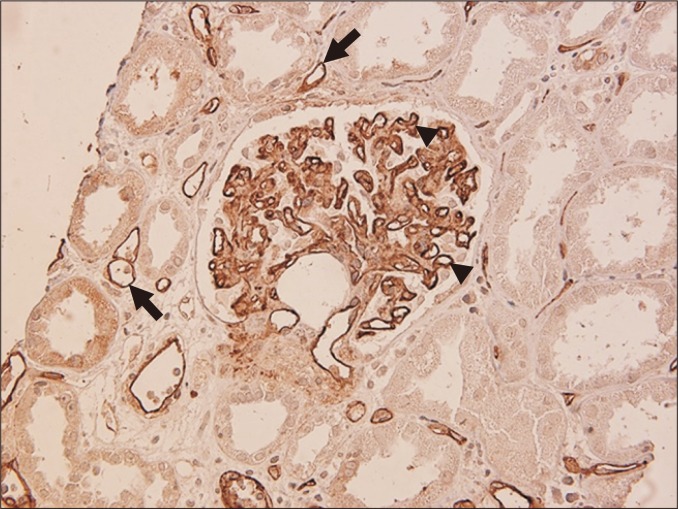
Glomerulitis is characterized by endothelial enlargement and inflammatory cell infiltration, often resulting in capillary luminal narrowing and destruction (Fig. 1). The infiltrating inflammatory cells may be T cells, monocytes, or neutrophils. This can be observed in the context of ABMR, and it is believed to be caused by endothelial injury that is mainly directed to human leukocyte antigen (HLA).
Inflammatory cells may also be present in non-ABMR conditions, such as acute T cell-mediated rejection (TCMR) and glomerulonephritis. For example, glomerular hypercellularity, which is referred to as ‘endocapillary hypercellularity’, is sometimes observed in immunoglobulin (Ig)A nephropathy that develops after transplantation [20]. This can therefore produce a diagnostic dilemma, particularly when the presence of concurrent ABMR is suspected.
Mesangiolysis
Mesangiolysis results from dissolution of the mesangial matrix and manifests as a pale area after periodic acid-Schiff (PAS) staining. It may be present in the context of ABMR, but may also occur with non-rejection conditions that are associated with endothelial or mesangial cell injury. The most common condition in which this occurs is thrombotic microangiopathy (TMA), but mesangiolysis may also be present in other glomerular diseases.
Mesangial matrix increase
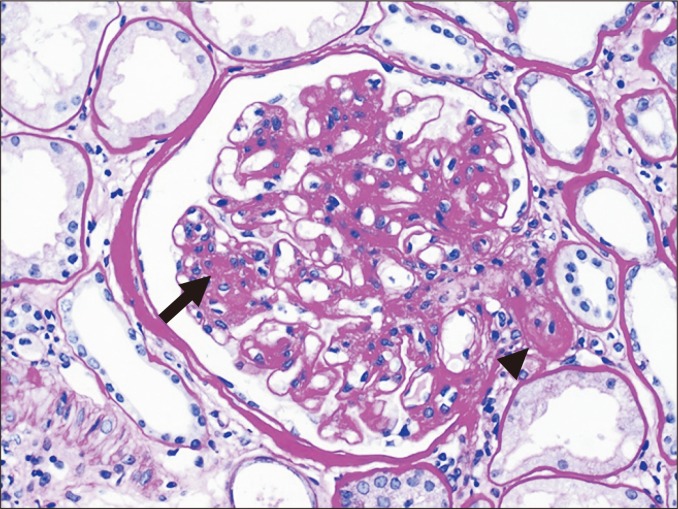
An increased mesangial matrix is defined as a matrix that exceeds the width of two mesangial cells in two adjacent glomerular lobules (Fig. 2). The mesangial matrix may be increased in association with chronic rejection; however, this feature is entirely nonspecific. In practice, increased mesangial matrix, along with mesangial hypercellularity, is frequently associated with IgA nephropathy or diabetic nephropathy post-transplant.
TMA
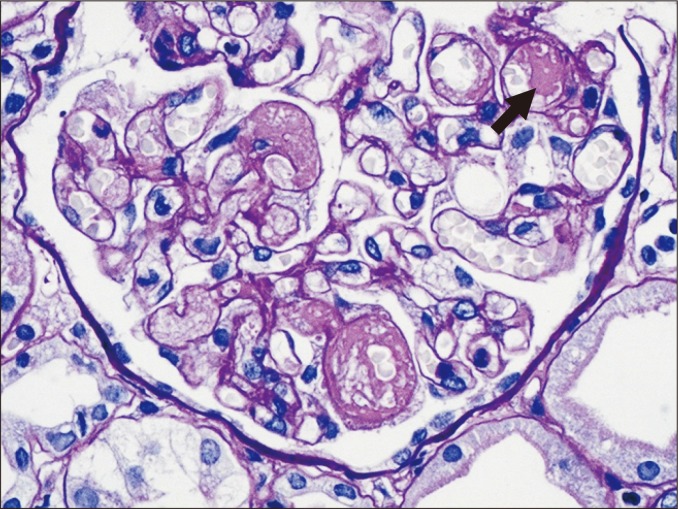
TMA is characterized by microthrombi, glomerular subendothelial electron-lucent widening, deposition of fluffy material, and the formation of a new subendothelial basement membrane (Fig. 3). This can be observed in the context of active ABMR related to endothelial injury. However, TMA may be present in other non-rejection conditions, such as recurrent atypical hemolytic uremic syndrome or drug-related conditions. In particular, calcineurin inhibitors, such as cyclosporine A and tacrolimus, induce dose-dependent endothelial dysfunction [21,22], and sirolimus, administered either alone or in combination with cyclosporine, can cause TMA [23]. Therefore, differential diagnosis may not be possible without both clinical history and laboratory data.
Transplant glomerulopathy
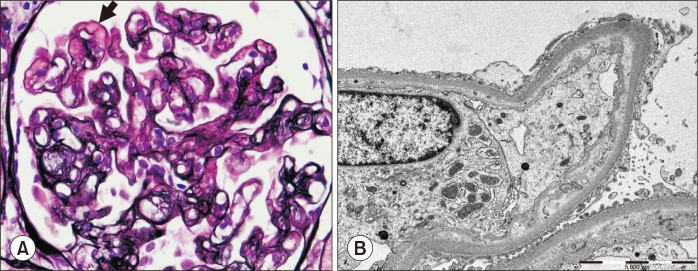
Transplant glomerulopathy is characterized by doubling or even multilayering of the glomerular basement membrane (GBM) (Fig. 4). GBM doubling is best detected with PAS or methenamine silver methods, but is only demonstrable by electron microscopy in the early stages of development. Transplant glomerulopathy may be seen in chronic ABMR that is caused by repeated endothelial injury and repair. Similar features can be present in other conditions, such as TMA and membranoproliferative glomerulonephritis associated with hepatitis C viral infection. However, glomerular immune complex deposition is a feature of membranoproliferative glomerulonephritis, whereas IgM and C3 deposits may be present in transplant glomerulopathy.
Tubules
Tubular degeneration/necrosis
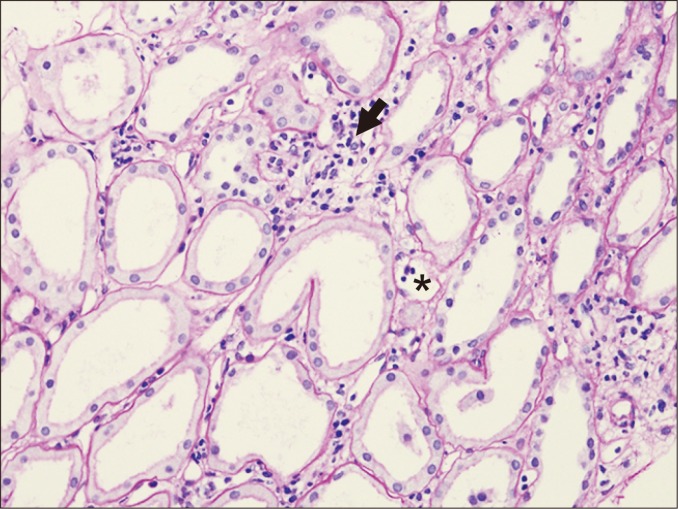
Acute tubular injury may be a sign of active ABMR if it is present in the absence of other known causes. Proximal tubular epithelial cells are most commonly affected, and flattening of the cytoplasm, loss of brush borders, and luminal dilatation can be observed (Fig. 5). Overt necrosis is not common, and intratubular microcalcification may be present.
Overt tubular necrosis is also often associated with polyomavirus infection. In this case, necrotic tubular cells may have intranuclear viral inclusions. In addition, acute tubular injury may be a sign of ischemia or drug toxicity. Therefore, observation of other clinical or immunologic features supportive of ABMR is required to confirm diagnosis.
Tubulitis
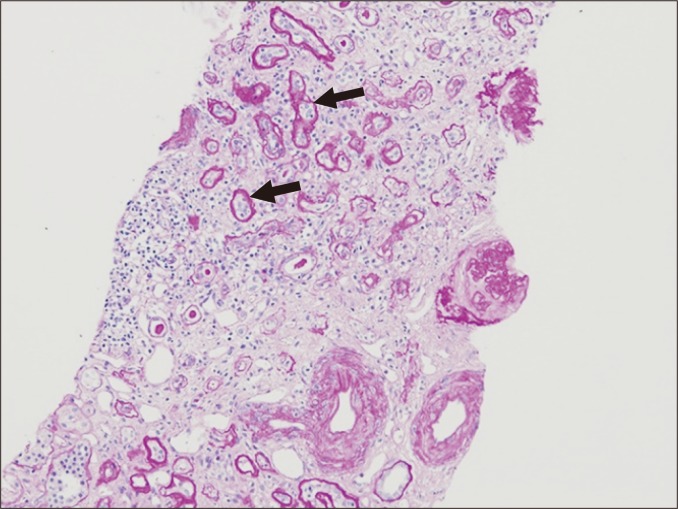
Tubulitis is a feature of acute and chronic active TCMR. It is characterized by the presence of inflammatory cells between tubular epithelial cells (Fig. 6). In acute TCMR, tubulitis is present in non-severely atrophic tubules. In chronic active TCMR, tubulitis involves both atrophic and non-atrophic tubules. The inflammatory cells present in tubulitis are most commonly lymphocytes and monocytes, but plasma cells may also be present.
Tubulitis can further result from infections and drug-induced interstitial nephritis, and it occasionally occurs in association with glomerulonephritis. Neutrophils may be dominant in bacterial infection, often with intraluminal neutrophilic abscess, and plasma cells are sometimes present in infiltrate from polyomavirus-associated interstitial nephritis. However, distinction between drug-induced interstitial nephritis and TCMR is not always possible.
Tubular atrophy
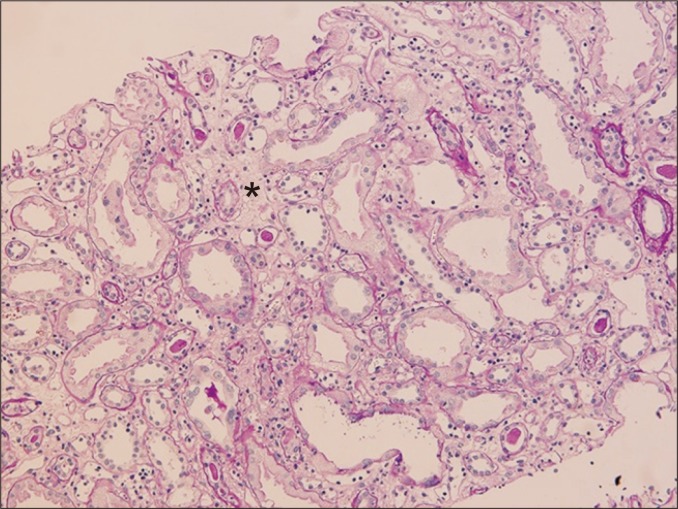
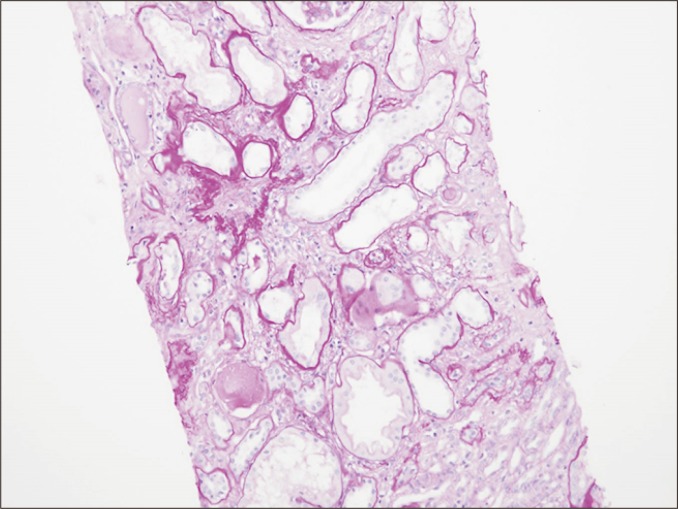
Tubular atrophy is a non-specific feature of chronic rejections and non-rejections. This is characterized by a > 50% narrowing of the tubular diameter or thickening of the basement membrane (Fig. 7). In some cases, this condition resembles endocrine glands without thickening of basement membrane. Severity of tubular atrophy is divided into three tiers (mild, moderate, and severe) based on the percentage of atrophic tubules (< 25%, 25% to 50%, and > 50%), and atrophic tubules are frequently replaced by interstitial fibrosis.
Interstitium
Interstitial edema
Interstitial edema is characterized by the presence of widened and lightly stained interstitium due to the accumulation of tissue fluid (Fig. 8). In acute TCMR, inflammatory cells are present in the edematous stroma.
Interstitial inflammation
Interstitial inflammation is a feature of acute and chronic active TCMR. In acute TCMR, inflammatory cell infiltration is present in the edematous stroma, associated with tubulitis of non-severely atrophic tubules (Fig. 8). In chronic active TCMR, inflammatory cells are found in both the edematous and fibrotic stroma, and this is accompanied by tubulitis of atrophic and non-atrophic tubules. The infiltrating cells are most often lymphocytes and monocytes, but plasma cells, neutrophils, or eosinophils can also be present. Immunohistochemistry has shown that T cells tend to infiltrate diffusely or in scattered patterns, whereas B cells are often aggregated. Inflammatory cells may also be present in Bowman’s capsule, resulting in Bowman’s capsulitis [24] in acute TCMR.
Notably, interstitial inflammation itself is a non-specific finding. Minimal inflammation may result from acute tubular injury and drug toxicity. In addition, interstitial inflammation can be present in drug-induced interstitial nephritis and polyomavirus-associated interstitial nephritis.
Interstitial hemorrhage
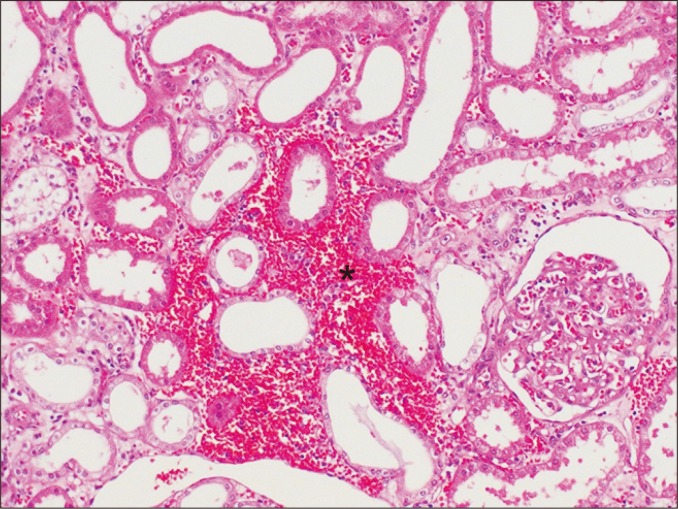
Interstitial hemorrhage occurs when extravasated red blood cells are present in the interstitium (Fig. 9). This may be seen in, but is not pathognomonic of, active ABMR or acute TCMR.
Interstitial fibrosis
In interstitial fibrosis, collagen fibrils and extracellular matrix components are increased in the interstitium (Fig. 10). This can be observed in both rejection and non-rejection conditions. In the context of chronic active TCMR, inflammatory cell infiltration may be seen in fibrotic interstitium, together with tubulitis of atrophic and non-atrophic tubules.
Blood vessels
Peritubular capillaritis
Peritubular capillaritis is characterized by the presence of both mononuclear and polymorphonuclear inflammatory cells in an often-dilated PTC lumen (Fig. 11). This condition is associated with active or chronic active ABMR. Although inflammatory cells may also be found in the PTC lumen in acute TCMR or other conditions, infiltration is usually mild and limited to areas of tubulointerstitial inflammation.
Arteritis
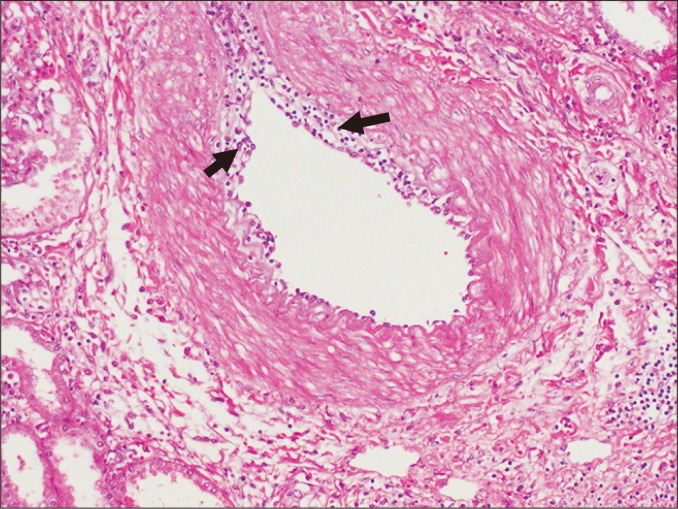
Arteritis can result from active ABMR and acute TCMR. Intimal arteritis is characterized by the presence of inflammatory cells, mainly T lymphocytes and macrophages, in the intima, often with lifting off endothelial cells (Fig. 12). In severe cases, transmural inflammation involving arterial media develops, accompanied by fibrinoid necrosis. If the intima does not fully recover to its original state, for example during chronic rejection, the intima will become thickened, forming neointima, which can trap inflammatory cells.
Multilayering of PTC basement membranes (PTCBML)
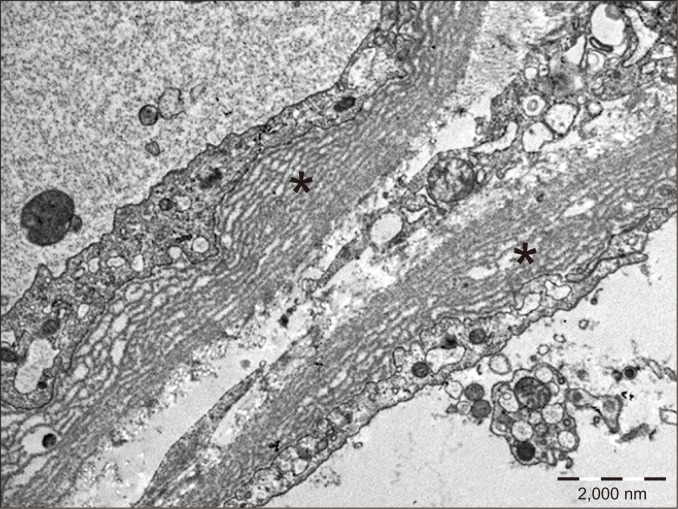
PTCBML is associated with chronic ABMR. This condition is usually detected by electron microscopy (Fig. 13), although thickening of capillary basement membranes can be seen by light microscopy in severe cases [25]. Similar to transplant glomerulopathy, PTCBML is believed to be the sequela of repeated PTC injury and repair, resulting in repeated basement membrane formation with progressive narrowing of the lumen. Severe PTCBML with circumferential involvement is more likely to be caused by alloimmune injury, whereas low-grade basement membrane layering with focal involvement is a frequent finding in non-rejections.
Transplant arteriopathy
Transplant arteriopathy can be caused by chronic ABMR and TCMR. In transplant arteriopathy, the arterial intima is thickened, forming a neointima (Fig. 14), which often contains foamy macrophages and lymphocytes. However, unlike fibroelastosis of hypertensive arteriosclerosis, elastic lamellation is not seen in transplant arteriopathy-associated neointima.
PTC C4d staining
Complement C4d is a product of complement activation, and linear, circumferential staining of this molecule in PTCs and the vasa recta (Fig. 15) can be used as a marker for ABMR. C4d binds to the endothelial surface via covalent bonding even after antibody has disappeared; therefore, it serves as a footprint for the antigen-antibody reaction. Glomerular C4d staining is more frequently observed in C4d-positive cases than in C4d-negative cases, although it has no clinical significance.
Evaluation of C4d positivity varies according to staining method. Immunofluorescence is more sensitive than immunohistochemical methods, and the staining intensity may also be affected by the length of time C4d was present at the antigen-antibody reaction site [26]. ABMR can occur without C4d positivity (C4d-negative ABMR), and C4d staining may be positive without rejection in ABO incompatible grafts.
Banff classification
The Banff classification for transplant pathology, which was first published in 1993 [8], has several important advantages: 1) It helps facilitate effective communication among transplant physicians, surgeons, and pathologists; 2) it is useful for assessing the effects of new immunosuppressive drugs in international multicenter clinical trials; and 3) it can be used for transplant research in addition to diagnosis.
The most recent Banff classification has six categories (Table 1) [27]. Rejection categories are Category 2 (antibody-mediated changes), Category 3 (borderline for acute TCMR), and Category 4 (TCMR). Before describing the different types of rejection, the basic requirements for biopsy interpretation and histologic scoring systems will be outlined.
Specimen adequacy
Requirements for specimen adequacy were established in Banff 1991 and Banff 1997. Currently, the presence of seven glomeruli with one artery is marginal, and ≥ 10 glomeruli with at least two arteries is considered adequate for numeric coding [9]. At least seven slides, three stained with hematoxylin and eosin (H&E), three stained with PAS, and one stained with trichrome stain should be evaluated.
Quantitative criteria for acute and chronic changes
Quantitative criteria for classifying acute and chronic changes in glomeruli, tubules, interstitium, and arteries/arterioles were established in 1991 and 1997 [9]. Only cortical tubules and interstitium are considered for evaluation.
Characters representing glomerular changes are ‘g’ (glomerulitis), ‘mm’ (mesangial matrix increase), and ‘cg’ (transplant glomerulopathy). Those for tubules are ‘t’ (tubulitis) and ‘ct’ (tubular atrophy), while for interstitium, they are ‘i’ (interstitial inflammation) and ‘ci’ (interstitial fibrosis). The ‘ti’ and ‘i-IFTA (interstitial fibrosis and tubular atrophy)’ characters, which indicate tubulointerstitial inflammation, were added in Banff 2007 and Banff 2015, respectively. ‘ti’ is defined as inflammation in total parenchyma, including scarred and non-scarred cortex [13], whereas ‘i-IFTA’ is defined as inflammation in areas of tubular atrophy and interstitial fibrosis [17].
For vessels, ‘v’ (intimal arteritis) and ‘cv’ (arterial fibrous intimal thickening) describe arteries, and ‘ah’ and ‘aah’ indicate hyaline arteriolar thickening [13]. In particular, although ‘aah’ was added to specifically address calcineurin inhibitor-related arteriolopathy, its specificity has not yet been verified. Two PTC criteria, ‘ptc’ and ‘C4d’, were added in Banff 2003 [11] and Banff 2001 [10], respectively, and refined in Banff 2007 [13]. ‘Ptc’ denotes peritubular capillaritis. ‘C4d’ is determined by either immunofluorescence or immunohistochemical methods, with different standards for positivity [13].
The Banff scoring system has three grades: mild (1), moderate (2), and severe (3). The cut-off points for ‘g’, ‘cg’, ‘mm’, ‘ct’, ‘i’, and ‘ci’ scores are < 25%, 25% to 50%, and > 50%. However, the cut-off points for ‘i’ are 10% to 25%, 25% to 50%, and > 50%, and for ‘ci’ they are 5% to 25%, 25% to 50%, and > 50%. Cut-off point for ‘t’ and ‘ptc’ scores are < 4, 4 to 10, and > 10 inflammatory cells/tubule or PTC [11,13]. Scores for ‘v’ are dependent on the extent of vascular wall involvement (< 25% intima, > 25% intima, and medial involvement), and for ‘cv’, they are dependent on the percentage of luminal narrowing (< 25%, 25% to 50%, and > 50%). Scores for ‘ah’ are dependent on the number and severity of hyalinosis. An asterisk (*) is added to express the type of inflammatory cells (‘i’, ‘ptc’), the presence of interstitial hemorrhage or infarction (‘v’), or arteriolitis (‘ah’) [27].
Antibody-mediated rejection
Diagnosis of active and chronic ABMR requires the presence of morphologic, immunohistologic, and serologic evidence. The morphologic features suggestive of active and chronic injury are unique, but active injury may coexist in chronic active ABMR. In contrast, the immunohistologic and serologic indicators are the same for active and chronic ABMR (Table 2) [27].
Although C4d was believed to be a distinct immunohistologic marker for ABMR [10,12], it was later recognized that C4d deposition can be present without morphologic evidence of active rejection [13], and C4d may be absent in ABMR [15,16]. Furthermore, donor-specific antibody (DSA) is not always detected in ABMR [28,29]. Therefore, ‘Increased expression of gene transcripts/classifiers strongly associated with ABMR’ was included as an alternative to C4d positivity in Banff 2017 [18].
The inclusion of ancillary molecular markers for diagnosis/differential diagnosis of ABMR has been supported by several reports that have described discrepancies between clinical, immunologic, and pathologic findings, as well as by the limitations of biopsy interpretation [30,31]. Sellarés et al [32] reported that microarray assessment of endothelial cells or NK gene expression was helpful in the diagnosis of ABMR. O'Connell et al [33], Modena et al [34], and Halloran et al [35] also showed benefits of gene expression profiling in the diagnosis of ABMR and identification of patients at risk of chronic injury. In addition, Reeve et al [36] demonstrated better prediction of graft survival from molecular scores than from histologic diagnoses.
T cell-mediated rejection
The diagnosis of acute and chronic TCMR largely depends on morphologic changes in the tubulointerstitial compartment. Diagnostic criteria for acute TCMR have not changed significantly since Banff 1991, and are graded according to active inflammation of non-atrophic tubules and interstitium and vessels. However, the criteria for chronic active TCMR were updated in Banff 2017 (Table 3) [27]. To diagnose chronic active TCMR, tubulointerstitial inflammation with a score > 2 should be present in areas of atrophic tubules and interstitial fibrosis, as well as in non-scarred tubules and interstitium [18,37-39].
Other revisions and Banff working groups
The term ‘CAN’ (chronic allograft nephropathy) was used to describe tubular atrophy and interstitial fibrosis, regardless of the underlying pathogenetic mechanisms, in the initial Banff 1991 report [8]. However, this term is problematic due to the fact that both rejections and non-rejections are included within the same group. It was therefore eliminated in Banff 2005 and replaced by specific diagnosis or the non-specific term ‘IFTA’ (interstitial fibrosis and tubular atrophy) [12].
Specialized Banff working groups have been convened to address issues relating to problematic areas in biopsy interpretation. The first working groups were organized at the Banff 2009 meeting, and these focused on ‘Isolated v-lesion’, ‘Fibrosis scoring’, ‘Polyoma virus nephropathy staging’, ‘Glomerular lesion scoring’, ‘Molecular pathology’, and ‘Quality assurance’ [14]. At the Banff 2011 and Banff 2013 meetings, the ‘C4d-Banff initiative for quality assurance in transplantation’ (BIFQUIT), ‘BK-BIFQUIT’, ‘Implantation biopsies’, ‘C4d-negative ABMR’, ‘TCMR’, ‘Clinical and laboratory assessment of highly sensitized patients’, and ‘Evaluation of adjunctive diagnostics in renal allograft biopsy interpretation’ groups were formed [15,16]. Working groups on ‘Electron microscopy’, ‘TMA’, ‘Recurrent glomerular disease’, ‘Composite surrogate end points’, ‘HIV+/HIV+ renal transplants’, and ‘Banff rules and dissemination’ were newly formed at the Banff 2015 and Banff 2017 meetings [17,18]. Several reports have been published from the working groups [40-42], and their efforts have been reflected in Banff updates.
Pitfalls of the current Banff classification
Although significant progress has been made in our understanding of rejection pathogenesis and in the refinement of morphologic and laboratory criteria for Banff classification, a number of uncertainties and drawbacks remain. These will be discussed in the following sections.
Criteria for quantitative scoring
In the Banff classification, glomerulitis is defined as endothelial cell enlargement along with the presence of inflammatory cells and capillary occlusion of ≥ 1 capillary loops. However, the number of inflammatory cells and minimum threshold for near-occlusion have not been clarified [43]. In addition, endothelial enlargement is subjective, and therefore, a mild degree of endothelial activation may not be recognized.
Presently, borderline changes are ‘suspicious’ for acute TCMR. The criteria for diagnosis of tubulitis and interstitial inflammation have also been changed at a number of meetings, resulting in confusion and inconsistent communication. For example, at Banff 2001, the criteria were ‘t1 and at least i1’. At Banff 2005 they were changed to ‘t1, t2, or t3 with i0 or i1’, and at Banff 2007, they were modified again to ‘t1, t2, or t3 with i0 or i1’ or ‘i2 or i3 with t1’. Finally, at Banff 2017, the criteria for tubulitis and interstitial inflammation were established as ‘t > 0 with i0 or i1’ or ‘i2 or i3 with t1’. In a recent report on borderline changes, these inconsistences have resulted in heterogeneous diagnostic groups [44].
Another parameter that may be considered for adjustment is PTCBML. This was defined as ≥ 7 PTC basement membrane layers in ≥ 1 PTC, or 5 to 6 layers in ≥ 3 PTCs. However, the minimum numbers and cut-off values of PTCBML are difficult to determine in daily practice [45]. Furthermore, electron microscopy, which is required for accurate diagnosis, is not available in many transplant centers.
Non-rejection conditions
Non-rejection conditions/diseases are grouped in Banff category 6 as a separate sheet [8]. However, with increasing graft survival, there have been increased opportunities to observe these conditions in late post-transplant biopsies, and they are often associated with graft dysfunction. For example, development of massive crescents in post-transplant IgA nephropathy or recurrent anti-neutrophil cytoplasmic antibody-associated glomerulonephritis may be a major contributor to graft dysfunction [46-48]. Additionally, recurrent focal segmental glomerulosclerosis can cause massive proteinuria. Therefore, integrated evaluation and interpretation of dominant injury patterns are required, particularly in cases with combined non-rejection conditions/diseases and rejections. Further, ‘g’, ‘cg’, and ‘mm’ scores are used in the classification of both rejection and glomerular diseases [49,50], but scores for crescents and global and segmental sclerosis, which are useful in the evaluation of glomerular diseases, are not included in the Banff parameters.
Uncertain pathogenesis for similar morphologies
The histology of intimal arteritis and transplant arteriopathy is rather distinct. However, it is difficult to determine whether the development of intimal arteritis and transplant arteriopathy is associated with cell or antibody-mediated mechanisms or if these conditions arose with or without rejection. One morphologic clue is the presence of other known features of TCMR or ABMR; however, this does not apply in cases of isolated vasculitis. Notably, inflammatory cell phenotyping in isolated vasculitis has revealed heterogeneity [51-53], which may reflect distinct injury mechanisms.
The observation of ‘C4d-positive renal transplant biopsies without evidence of rejection’ is also associated with uncertainty. Intriguingly, one study performed gene expression analysis and identified patients at higher risk of developing ABMR among these cases [54].
Others
In the current Banff classification, interstitial inflammation is defined by its extent. However, there is no way to determine whether inflammatory cells are active or quiescent or if their composition is related to inflammatory activity or fibrosis. Further, asterisks in some Banff scores, some related to inflammatory subtypes, are not popular, and their significance is not known.
Future perspectives
The discovery and application of non-invasive molecular markers for effective diagnosis of renal transplant rejection are active fields of research [55-57]. Importantly, to develop a comprehensive approach, integration of tissue pathology assessment, transcriptomics, and urinary proteomics may be needed, particularly in cases of chronic TCMR [58]. Recently, the NanoString Banff consortium was launched to develop the nCounter Human Organ Transplant Panel to identify biomarkers of rejection, uncover the mechanisms behind tissue damage, and monitor immunosuppressive drug toxicity and infections using paraffin-embedded renal tissue [59]. Diagnosis of mixed rejection and the often problematic coexistence of polyomavirus nephropathy and rejection or calcineurin inhibitor toxicity and rejection may benefit from this approach. Furthermore, clinical significance of antibody responses to non-HLA antigens, such as major histocompatibility complex class I chain-related proteins A and B and angiotensin II type 1 receptor, may be clarified [60,61].
Immunophenotyping of allograft biopsies may also provide some additional benefits, for example facilitating the detection of glomerulitis. In particular, this strategy may help us to understand and distinguish the inflammatory and profibrotic phases of inflammation and/or the activity of inflammation. To this end, multiplex immunohistochemistry is likely to be especially useful, as inflammatory cells are mixed in nature and vary throughout the rejection process [62-64]. Digital pathology has become increasingly popular in pathology practice, both in clinical biopsy diagnosis and in research. This strategy has the potential to reduce inter-observer variation and increase objectivity through use of quantification. For example, interstitial inflammatory phenotypes have been reported from digitization of immunostained biopsy sections and algorithm-driven analysis, with good clinical correlation [65]. Deep learning-based histopathological assessment of transplant biopsies [66] has also been performed, and high correlations between the neural network’s quantification of glomeruli, interstitium, and atrophic tubules and results from renal pathologists were reported.
As noted previously, addition of scores for crescents and global and segmental sclerosis to the current Banff scores is necessary for integrated evaluation of biopsies from patients with combined non-rejection conditions/diseases and rejections.
Lastly, with recent advances in bioengineered organs, we may also need to implement strategies to deal with abnormalities in bioengineered organs. Recently, Solez et al [67] proposed the construction of a framework for the classification of tissue engineering pathology at future Banff Transplant Pathology meetings, in collaboration with the Human Cell Atlas project [68].
To summarize, for precision diagnosis of transplant rejection, integration of histological, laboratory, and tailored molecular markers is necessary. Artificial-intelligence based assistance has the potential to further improve diagnostic accuracy.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









