


Introduction
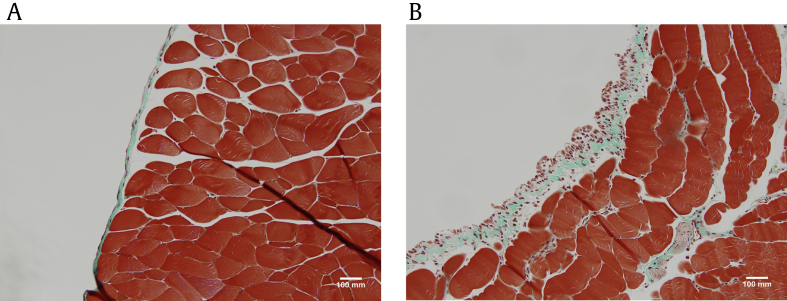
Peritoneal dialysis (PD) is a widely used renal replacement therapy for patients with end-stage renal disease [1], [2]. The healthy peritoneum is a semipermeable membrane consisting of a superficial mesothelial layer, a basement membrane, and a thin submesothelial zone (Fig. 1A) [3]. Long-term exposure to bioincompatible dialysis solutions, uremia [4], and intermittent peritonitis [5] leads to a low-grade chronic inflammation, triggering a reparative response. This response normally provides restoration of tissue structure with minimal loss of function. However, this reparative mechanism may become dysregulated. This results in tissue fibrosis with deposition of extracellular matrix (ECM), angiogenesis, and eventual membrane failure (Fig. 1B). The appearance of myofibroblasts in the submesothelial tissue is a critical component of peritoneal membrane fibrosis and angiogenesis [5].
The myofibroblast in fibrosis
The myofibroblast is a specialized contractile cell that expresses both fibroblast and smooth muscle cellâlike characteristics [6]. Myofibroblasts are motile cells and therefore possess stress fibers and ruffled cell membranes due to changes in the actin cytoskeleton [7]. Stress fibers are composed of bundles of actin microfilaments and contractile proteins. Actin microfilaments terminate at the cell surface in a specialized adhesion complex termed the fibronexus that becomes linked to extracellular fibronectin fibrils [7]. This creates a mechanotransduction system that allows for transmission of force from stress fibers to the ECM. Similarly, mechanical signals from the extracellular environment can activate intracellular signaling pathways [8]. Expression of alpha smooth muscle actin (α-SMA) is a key marker that distinguishes the myofibroblast from the fibroblast [9]. Although the function of the myofibroblast has not fully been elucidated, the myofibroblast has mostly been studied for its role in wound healing and its presence during pathological conditions such as fibrosis. Originally, myofibroblasts were observed in granulation tissue of healing wounds. They were observed to produce the contractile force required during wound contraction. More recently, several studies have identified the myofibroblast as an important element in the production of ECM and progression of fibrosis [7], [10].
In a healthy environment, stromal cells including fibroblasts and perivascular cells remain quiescent, producing little ECM and few actin interactions between cells and cell to matrix [8]. In response to injury, myofibroblasts accumulate at the site of injury where they produce ECM in response to cytokines from local cells [6], [8]. Transforming growth factor beta (TGF-ÎČ) is a key cytokine that regulates expression of α-SMA and type 1 collagen in the myofibroblast [9], [11]. Moreover, the differentiation process of the myofibroblast is largely regulated by TGF-ÎČ. In addition to TGF-ÎČ, myofibroblasts are also stimulated by changes in the mechanical microenvironment and high extracellular stress [7], [8]. During normal conditions, the ECM maintains its cross-linked structure; however, tissue injury results in remodeling of the ECM and changes in the mechanical environment. In response to this change in ECM structure and TGF-ÎČ signaling, precursor cells begin to acquire contractile stress fibers [7]. During this time, myofibroblasts express some features that are similar to fibroblasts such as actin stress fibers. This transitional cell has been characterized as the protomyofibroblast [8], [12]. The protomyofibroblast begins to acquire more contractile activity with the expression of α-SMA in the stress fibers and can now be characterized as a myofibroblast. ECM stiffness is one promoter of myofibroblast maturation [12], [13]. During the final stages of wound healing, the ECM regains its normal structure and the myofibroblasts undergo apoptosis [8], [12].
The last component of the wound-healing response involves replacement of injured cells with new cells and resolution of scar tissue to preserve functional and structural integrity of the organ [14]. However, this is not always the case depending on the type and duration of injury as well as the severity of damage. Often, chronic injury may result in the inability to replace scar tissue with functional tissue resulting in irreversible fibrosis [14]. Therefore, fibrosis is largely characterized as accumulation of ECM including collagen, proteoglycans, and fibronectin [10]. The myofibroblast is the primary effector cell that is activated during this process [10]. Myofibroblast populations have been noted in abnormal wound healing in several vital organs such as the kidney, lung, liver, and heart [11], [15], [16].
Myofibroblasts have also emerged as the main effector cell contributing to peritoneal fibrosis. The presence of the myofibroblast contributes to the loss of integrity of the peritoneal membrane in long-term PD patients by inducing collagen deposition resulting in structural and functional changes in the peritoneal membrane [17]. In PD patients, the myofibroblast is dominant in the peritoneal membrane even before fibrosis is present in contrast to the normal healthy peritoneum [18].
It is challenging to study stromal cells; fibroblasts do not express any specific markers resulting in difficulties distinguishing them from other cells [10]. Several studies have used fibroblast specific protein (FSP) 1 (also known as S100A4) as a fibroblast-specific marker [19]. Recent evidence demonstrates FSP1 is not specific to fibroblasts and is also expressed in other cells such as monocytes [40]. Myofibroblast expression of α-SMA is increasingly being used as the method of identifying the myofibroblast. However, this marker still presents some challenges as α-SMA is also expressed by other cells of the mesenchymal lineage such as vascular smooth muscle cells [10].
Origin of the myofibroblast: current hypotheses
As the myofibroblast is the key effector cell in progression of fibrosis, it is of scientific and practical interest to elucidate the origin of this cell. This is a controversial area of research. In various research studies, myofibroblasts have been shown to derive from different cellular sources including resident fibroblasts [8], [16], epithelial cells via epithelial to mesenchymal transition (EMT) [20], endothelial cells (endothelial to mesenchymal transition) [21], perivascular cells [22], and bone marrowâderived cells [19], [23]. Interestingly, the original hypothesis was that myofibroblasts arose from locally residing mesenchymal cells such as resident fibroblasts and pericytes/perivascular fibroblasts [8], [16] based on studies of dermal wound healing [12], [24] (Fig. 2A). This was also supported in models of fibrosis such as scleroderma [25] and liver and renal fibrosis [12], [26]. However, this view quickly changed as a result of accumulating evidence in support of conversion of local epithelial cells via EMT [27] (Fig. 2B). The first piece of evidence comes from studies in vitro where epithelial cells can be transformed into myofibroblasts by exposing cells to wound-healing cytokines such as TGF-ÎČ [27]. Secondly, evidence from experimental fibrosis in animal models has provided us with snapshots of molecular markers of EMT in vivo
[28]. Myofibroblasts appear in the submesothelial zone in experimental models of peritoneal fibrosis [17] and have been observed in biopsy samples in PD patients with peritoneal fibrosis [29]. The origin of the myofibroblasts in peritoneal membrane injury remains an area of active research. Early studies, including our own work, have pointed to EMT as a source of submesothelial myofibroblasts. More recently, these observations have been challenged by cell fateâtracing experiments [30]. There is also some experimental evidence that myofibroblasts can originate from the bone marrow and play a role in peritoneal fibrosis [19], [23].
In vitro and ex vivo evidence for EMT
The study of the origins of the myofibroblast began in vitro and moved into experimental animal models. Initial evidence supporting EMT comes from studies in vitro where epithelial cells have been induced to transform into myofibroblasts during conditions of injury [31], [32]. This process has been extensively studied in renal fibrosis where primary tubular epithelial cells exposed to TGF-ÎČ undergo EMT [33].
These studies have been fundamental in our understanding of the EMT process by defining the basic markers used to characterize EMT and the signaling pathways involved during this process [28]. EMT can be considered as a 2-step process, the primary step being cellular transition followed by invasion into the interstitial tissue (Fig. 2B) [27]. In vitro studies have outlined the process of cellular transition, but in standard 2-dimensional cell culture, cellular invasion has been more difficult to study; EMT is characterized by the loss of cell adhesion through downregulation of intercellular adhesion molecules such as E-cadherin, loss of cell polarity, rearrangement of the cytoskeleton with an increased expression of α-SMA, and acquisition of migratory behavior (Fig. 2B) [27], [28]. During the last stage of EMT, the basement membrane can be degraded and the mesenchymal cell can migrate into the interstitial layers and deposit collagen resulting in fibrosis (Fig. 2B). Metalloproteinases (MMPs) are usually thought to be involved in degradation of the basement membrane and may facilitate invasion of transitioned cells [27], [34].
The EMT cellular program is driven by initiating factors such as hypoxia, inflammation, or growth factors such as TGF-ÎČ. These activate transcriptional regulators such as Snail, Zinc finger E-box-binding homeobox, or Twist [35]. These transcriptional regulators then initiate a genetic program that leads to the EMT phenotype [35].
Studies in vitro have been useful because they can demonstrate the transitional aspect of EMT by measuring cell movement and combining this with changes in cell phenotype [36]. Matrigel (Corning Life Sciences, Tewksbury, MA) is a common substrate used to measure cellular invasion, and studies have used this as a method of measuring cell movement [37]. However, cross-linked, mature basement membrane may not be susceptible to protease activity as is Matrigel [38]. Similarly, collagen invasion assays have been used to study cell invasion against laminin, type I collagen, and type IV collagen. However, in vivo, the triple-helical structure of collagen is resistant to proteolytic attack [38]. Therefore, analysis of invasive EMT is difficult to detect in vivo, and investigators usually have to rely on recognition of certain markers that reflect transition [36].
EMT was first demonstrated in the human peritoneum after dialysis in a landmark paper in 2003 by Yåñez-MĂł et al [39]. An ex vivo culture of mesothelial cells was developed from peritoneal effluent from patients on PD. They found that the cell morphology could be classified as epithelioid like or nonepithelioid like. The epithelioid cells demonstrated the usual markers such as cytokeratin, E-cadherin, and intercellular adhesion molecule 1. There was decreased E-cadherin and cytokeratin expression in cells with a fibroblast phenotype compared with those with a more epithelioid phenotype [39]. The epithelial markers also declined with time spent on PD. The gene expression of Snail was increased in transitioning and fibroblast like cells grown from peritoneal effluent. Confluent layers of mesothelial cells were also mechanically wounded, and fibroblastic cells appeared in the layer of migrating cells near the wound. Cultured cells treated with TGF-ÎČ displayed decreased amounts of E-cadherin and increased levels of Snail. Biopsy specimen taken from 9 PD patients confirmed the results from the ex vivo mesothelial culture [39]. These results have been confirmed by a similar study of mesothelial cells also derived from peritoneal effluent [2]. These studies strongly suggest that myofibroblasts may originate from injured mesothelial cells undergoing EMT.
These ex vivo cell-based experimental results have been questioned by recent work in a rodent model of dialysate exposure [40]. Cho et al [40] found that the evaluation of EMT markers in the cells grown from the peritoneum of rats treated with PD solutions did not correlate well with the EMT observed in the peritoneal tissues. Whether these results in a rat model can be applied to patients on PD is still an open question.
Animal models to study peritoneal membrane injury
Experimental animal models can recreate aspects of a pathophysiological condition and can thus help to identify mechanisms of disease and identify targets for therapeutic intervention. The extent to which an animal model of human disease actually simulates the disease state is always open to criticism [41].
Different animal models have been developed to study the underlying challenges in PD including viability of the peritoneal membrane and biocompatibility of dialysis solutions [42]. Experimental animal models give us the ability to study mechanisms underlying peritoneal membrane injury and could lead to improvements in current techniques. Historically, investigators have used rat or rabbit to model PD, generating acute and chronic models of PD. In a standard acute model of PD, fluid is infused by intraperitoneal injection or a temporary catheter into the peritoneal cavity in mice, rats, rabbit, and sheep [43], [44]. This model has been important in understanding properties of solute transport across the peritoneal membrane, changes in the interstitial matrix, and changes in attachment and morphology of mesothelial cells during injury [[45], [46], [47]]. The chronic infusion model of PD uses a peritoneal catheter implanted into rodents which then receive a daily or twice daily infusion of dialysis fluid for a span of 4â20 weeks [48]. Although this model closely mimics the progression of peritoneal membrane injury, the indwelling catheter has been shown to amplify the inflammatory response via a foreign body reaction [49].
The model used extensively in our laboratory and others involves gene transfer of TGF-ÎČ into the rodent peritoneum [50]. TGF-ÎČ is one of the major mediators of peritoneal fibrosis and angiogenesis and can be delivered to the peritoneum to model this condition. Adenovirus-mediated gene transfer of TGF-ÎČ has been used to model peritoneal fibrosis in mice [50] and rats [51]. This model has demonstrated changes in the functionality and structure of the peritoneum similar to what is seen in patients on long-term PD. We also have a helper-dependent adenovirus expressing TGF-ÎČ to induce prolonged expression of the transgene, and this resulted in changes in the peritoneal structure as observed in encapsulated peritoneal sclerosis (EPS) [52]. The helper-dependent adenovirus allows for longer duration and lower expression of transgene with an attenuated immune response [52].
EPS is a rare disease that occurs in a small minority of PD patients [52]. Peritoneal fibrosis may be an underlying factor in the development of EPS, and therefore, the study of EPS can also provide clues to understanding peritoneal fibrosis. One of the most commonly used agents to model EPS in animals is chlorhexidine gluconate [53]. Chlorhexidine was initially used as a topical antiseptic agent to prevent peritonitis but became associated with an increased risk of EPS and is now used as an experimental model of EPS. Daily intraperitoneal injections of chlorhexidine gluconate can induce peritoneal fibrosis and angiogenesis in mice and rats [54].
Animal models and EMT
Several animal models of peritoneal membrane injury have been generated which support the hypothesis that the epithelium is a source of myofibroblasts. Supported by extensive in vitro data for EMT, researchers have studied common peritoneal membrane injury models to confirm EMT in vivo
[2], [17]. The identification of in vivo EMT involves use of surrogate markers to identify changes in expression of epithelial markers, cell-to-cell adhesion molecules, cytoskeletal changes, and altered regulation of transcription factors associated with EMT [28], [55]. Some of the major and most common markers used to identify EMT include loss of epithelial markers including E-cadherin and cytokeratin and gain of mesenchymal markers such as α-SMA and Snail [55]. The most common method that has been used to provide better visualization of the EMT process is immunofluorescence analysis of epithelial and mesenchymal markers [28]. Cells undergoing transition can then be identified by analyzing the co-localization of both epithelial and mesenchymal markers in cells in the peritoneal membrane [55]. However, these studies have received some criticism as static markers of EMT are commonly used. Changes in these markers may not be representative of EMT as EMT is a more transitory process [28]. Detecting EMT in vivo has presented several limitations. As mentioned previously, there are no specific markers for myofibroblasts, so immunohistochemistry is confusing because of nonspecificity [55]. EMT is a reversible process, so there may be stages where cells revert to their parental phenotype and lose their mesenchymal characteristics [55]. There are also more advanced stages where epithelial cell characteristics are lost completely. Therefore, the detection of transitioning cells critically depends on the timing of detection after peritoneal membrane injury [55]. Detection of intermediate stages of EMT in injured tissue is a simple method that has been used in the past to identify transitioning cells in vivo
[55]. Intermediate stages of EMT are characterized by cells expressing epithelial characteristics such as cytokeratin and E-cadherin but have also acquired mesenchymal markers such as α-SMA. Therefore, these cells co-express epithelial and mesenchymal features at the same time [20] (Fig. 3).
Aroeira et al [2] used these immunohistologic methods to identify EMT in vivo using an animal model of exposure to glucose-based PD fluids to induce peritoneal fibrosis and angiogenesis over a period of 5 weeks. This exposure was accompanied by a failure in ultrafiltration. After 2 weeks of PD fluid exposure, there was a loss of mesothelial cells from the peritoneum. This closely mimics the structural changes that occur in patients on PD [2]. Immunofluorescence staining for both an epithelial marker, cytokeratin, and the myofibroblast marker, α-SMA, was used to identify mesothelial cells and myofibroblasts, respectively. Dual-labeled cells expressing both cytokeratin and α-SMA were considered cells undergoing EMT in the intermediate stages. This analysis revealed that the loss of mesothelial cells from the peritoneal membrane surface coincided with the appearance of cells labeled with cytokeratin and α-SMA in the submesothelium [2]. We have also examined the process of EMT by combining analysis of EMT-related genes and proteins along with immunofluorescence staining for EMT markers in our model of adenovirus-mediated TGF-ÎČ gene delivery (Fig. 3). TGF-ÎČ induces peritoneal fibrosis and angiogenesis in the rodent peritoneum, creating a robust model of peritoneal membrane injury [17]. Rat peritoneum exposed to adenovirus expressing TGF-ÎČ exhibited increased submesothelial thickness and angiogenesis. We observed an early increase in expression of Snail, α-SMA, MMP-2, and type I collagen. Laminin staining revealed a disruption of the basement membrane at an early time point. Dual staining revealed cells expressing both epithelial and myofibroblast characteristics in the submesothelial layer [17]. In this study, along with other studies done in vivo, submesothelial α-SMA-positive myofibroblasts without epithelial markings are of an unknown origin. They may be fully transformed epithelial cells, transformed fibroblasts, or other cell types recruited locally or from the circulation. Therefore, these experiments suggest that at least some myofibroblasts in the submesothelial space may come from mesothelial cell conversion and support the notion of EMT-derived myofibroblasts.
The use of immunofluorescence labeling of epithelial and myofibroblast markers to identify cells undergoing EMT has been criticized. This provides only a static picture of the cellular response to injury, and we have shown that this picture looks very different depending on the point of time in the injury response [33], [45]. Furthermore, there are criticisms of immunofluorescence studies including the nonspecificity of antibodies used and image-processing artifacts. Different models of peritoneal injury may yield different temporal patterns of damage and may thus make detection of EMT challenging. For example, the adenovirus-mediated gene transfer model of TGF-ÎČ yields a fairly rapid fibrogenic response, whereas daily exposure to PD fluids will lead to a more gradual onset of fibrosis, and it will likely be more difficult to detect EMT using dual immunofluorescence studies. Finally, different animal strains may have very different response to fibrogenic stimuli [35].
We have quantified the EMT response and find that very few cells express both mesenchymal and epithelial markers in the peritoneum after injury. In general, we find only 1â2 cells per high power field to be dual labeled [50], [56]. This could indicate that EMT is a rare event in peritoneal fibrosis or that our âsnapshotâ of peritoneal membrane injury captures only those cells in the process of transition, not those cells fully transitioned to a myofibroblast phenotype.
Fate-tracing experiments and evidence refuting EMT in peritoneal fibrosis
An alternate method of visualizing the origin of cells in a complex injury response system has been developed using inducible genetic fate mapping. Emerging evidence from these studies sheds more light on the origin of the myofibroblast in tissue injury and throws into question the dominant role of EMT in the appearance of submesothelial myofibroblasts. Genetic fate mapping is a technique that allows researchers to detect the origin of cells with molecular precision [16], [28]. Mammalian fate-mapping studies have adopted the LoxP/Cre recombinase system. A mouse is constructed where a reporter molecule such as green or red fluorescent protein (GFP or RFP) is inserted in the genome. This reporter gene is silenced with an upstream stop codon that is flanked by LoxP sites (floxed) [16]. When this mouse is mated with a mouse that has a tissue-specific promoter driving Cre recombinase, the stop codon in that tissue is permanently removed, allowing for permanent expression of the reporter. The label is driven by a strong, constitutively active promoter. Therefore, all descendent cells will express this reporter molecule. Moreover, this system can be modified to generate inducible reporter expression or tissue-specific reporter [16]. For example, in the kidney, podocyte-specific gene expression can be achieved by crossing reporter mice with a podocin promoterâCre gene construct. Cre will be expressed in a tissue-specific manner, resulting in permanent labeling of podocytes. This system can be manipulated further by using an inducible promoter for the Cre gene. That way, Cre can be turned on in a cell-specific manner by adding an inducing agent, such as tamoxifen for an estrogen receptorâinducible promoter [16].
Peritoneal mesothelial cells are somewhat more difficult to label as a specific marker is not available. The closest available is a Wilms tumor-1 (WT1) promoter that labels mesothelial cells and developing podocytes. Recent studies by Chen et al [30] using this system will be outlined in the following paragraphs.
Earlier work using lineage tracing came from models of kidney injury and fibrosis. Humphreys et al [22] used several different kidney-specific promoter mice to demonstrate that myofibroblasts do not develop from injured tubular epithelial cells in vivo; however, the in vitro response was similar to earlier reported studies [17]. They did arrive at the surprising result that the majority of interstitial myofibroblasts actually came from perivascular cells within the interstitium after kidney injury [22]. These striking results contradicted many previous studies, suggesting that EMT was a source of interstitial myofibroblasts after kidney injury.
Similar to Humphreys et al [22], LeBleu et al [57] also used genetically modified mice to track the origin and function of the myofibroblast. Mice expressing yellow fluorescent protein driven by the α-SMA promoter mediated by the LoxP/Cre system in a unilateral ureteric obstruction model of renal fibrosis exhibited an accumulation of α-SMA+ myofibroblasts in the interstitium of fibrotic kidney [57]. Bone marrow transplant experiments using α-SMA-RFP mice and immunolabeling for α-SMA+ cells revealed that 35% of myofibroblasts in the interstitium of injured kidneys originated from the bone marrow and 65% originated from resident cells including pericytes. Interestingly, ablation of bone marrowâderived myofibroblasts reduced kidney fibrosis, whereas the deletion of pericyte-derived myofibroblasts did not [57]. Using mice expressing yellow fluorescent under the control of the Îł-glutamyl transferase promoter (ÎłGT) crossed with α-SMA-RFP mouse, LeBleu et al [57] were able to demonstrate that a small number (about 5%) of interstitial myofibroblasts in the kidney arose from epithelial cells through EMT.
Similar studies have been recently carried out by Chen et al [30] in models of peritoneal fibrosis. Transgenic mice were generated to conditionally express red fluorescent protein in WT1+ cells. The WT1 promoter was conditionally induced using tamoxifen to label mesothelial cells in adult mice. These authors used several models of peritoneal membrane injury including hypochlorite injection, exposure to dialysis fluid, and adenovirus-mediated gene transfer of TGF-ÎČ [30]. They observed that WT1 labeled the majority of mesothelial cells along with an occasional submesothelial cell of unknown type. Following injury, very few WT1+-RFP cells co-expressing α-SMA were found in the thickened laminin scar. WT1+-RFP cells remained in the mesothelial layer. Type I collagen (Col1A) reporter mice were used to label collagen-producing cells in the peritoneum. α-SMA/Col1A-positive cells were found in the interstitium as myofibroblasts [30]. These were subsequently shown to originate from existing submesothelial fibroblasts. Peritoneal mesothelial cells expressed Col1A after injury, but not α-SMA, suggesting that the mesothelium participates in the injury response, but not through EMT [30]. Because this study is one of the first studies in the peritoneum to directly contradict EMT as an origin of myofibroblasts, confirmatory studies would be welcome. Furthermore, the expression of WT1 in the submesothelium is not as specific a cellular marker for mesothelium as kidney-specific markers. The inducible WT1 promoter did not label all mesothelial cells but did label a proportion (15%) of submesothelial fibroblasts [30]. The fibroblast origin of myofibroblasts in this study was confirmed using a double transgenic Col1A1 reporter mouse with an inducible Col1A2 reporter construct. However, 65% of submesothelial fibroblasts were labeled with Col1A2, and these expanded to become roughly 65% of all myofibroblasts [30]. Therefore, the cell-specific markers in the peritoneum are neither completely specific nor sensitive. This may have an impact on the results obtained. Finally, in all these lineage-tracing experiments, it is theoretically possible for the lineage promoter to become transcriptionally silenced [28].
What is the role of epithelial cell transition in organ fibrosis?
Studies of myofibroblasts in organ fibrosis, including peritoneal fibrosis, have focused on the origin of these cells in the interstitial or submesothelial tissue. It is becoming increasingly clear, however, that the injured epithelium may play a critical role in directing organ repair without undergoing a full EMT process with transition and migration to the interstitium. Strong evidence for this comes from a recent article by Lovisa et al [58] in evaluation of the role of the epithelium in directing renal fibrosis. These researchers developed a mouse model where the key EMT transcription factors Twist or Snail were silenced specifically in the renal tubular epithelium. In a model of renal interstitial fibrosis, they demonstrated that silencing these transcription factors blocked the expression of α-SMA in the renal tubular epithelium. By preventing EMT, or at least cellular transition of epithelial cells, the extent of the renal interstitial fibrosis was abrogated [30].
In the work by Chen et al using a Col1A reporter mouse, the injured mesothelial cells began to express collagen after peritoneal injury. They did not identify α-SMA staining of these cells [30]. In our own models, we have seen several instances where the peritoneal mesothelium undergoes a cellular transition and begins to express both cytokeratin and α-SMA, without migration of these cells to the interstitium (in situ EMT) [34] (Fig. 2C). Using laser capture microdissection, injured peritoneal mesothelial cells expressed Snail, α-SMA, and vascular endothelial growth factor, suggesting these cells undergo EMT and elaborate growth factors that induce submesothelial resident cells to participate in the wound-healing response [59]. The mesothelium is maintained in a healthy balance by a combination of profibrotic and antifibrotic factors. Among the antifibrotic factors is bone morphogenic protein 7 which has been shown to be antagonized by the secreted factor Gremlin [60]. Recently, we overexpressed Gremlin in the peritoneum, which led to in situ EMT. This cellular transition alone was able to drive the subsequent peritoneal fibrogenic response with matrix deposition and angiogenesis [56].
Therefore, recent evidence concerning the origin of myofibroblasts in organ fibrosis using cell fateâtracing experiments may not be as contradictory with previous research as initially thought. Our previous work is consistent with the possibility that few of the myofibroblasts in the interstitial tissue are derived from the overlying epithelium. Despite this, there is increasing evidence that injured epithelial cells undergo a limited transition, or in situ EMT, and this may be sufficient to direct the subsequent interstitial or submesothelial fibrosis.
Summary
The origin of the myofibroblast requires further research as the recent evidence from fate-mapping studies contradicts evidence from the past [22], [30]. This could be attributed to technical difference between techniques and studies, or it could be related to the development of the disease process and timing of pathogenesis. Research from in vitro studies of the origin of the myofibroblast has been useful in identifying surrogate markers of EMT including the loss of epithelial markers, E-cadherin, and cytokeratin and gain of mesenchymal markers, α-SMA, and Snail. Using these surrogate markers, several animal models of peritoneal membrane injury have provided evidence in support of EMT as a source of myofibroblasts [55]. Most commonly used, immunofluorescence methods have identified the transitioning cells in vivo by analyzing co-localization of epithelial and mesenchymal markers [28]. Moreover, most studies in vitro and in vivo support the injured mesothelial cell as the progenitor of the myofibroblast, and there is one recent study with opposing results [30]. Studies of fibrosis in other organs also present evidence in support of EMT [27]. Recently some studies of renal fibrosis present results opposing epithelia as a major source of the myofibroblast but still suggest that a smaller portion of cells derive from EMT [17]. Therefore, confirmatory studies are necessary to further understand this process and to understand to what extent EMT is contributing to the myofibroblast population. This may be explained by the concept of in situ EMT. In other words, injured epithelial cells may not fully transition to yield a complete EMT, and cells may only transition to an intermediate stage. A more specific method is required to define and identify EMT as well as account for the disease model that is being used to truly understand its contribution to the origin of the myofibroblast. In the end, this may require using a combination of lineage-tracing studies and EMT criteria. The role of in situ EMT in driving peritoneal fibrosis could be further evaluated by deleting key EMT transcription factors, such as Snail or Twist, in the mesothelium in a model of peritoneal fibrosis. Elucidating the origin of myofibroblast and better defining the role of the mesothelium will lead to development of new antifibrotic therapies and improvement of current treatment methods.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









