Successful diagnosis and treatment of recurrent atypical hemolytic uremic syndrome posttransplantation caused by the heterozygous deletion of CFH in a patient with end-stage kidney disease of uncertain etiology
Article information

Atypical hemolytic uremic syndrome (aHUS) is a rare disease caused by the dysregulation of the alternative pathway of the complement system, leading to microvascular damage. It is a type of thrombotic microangiopathy (TMA) characterized by non-immune hemolytic anemia, thrombocytopenia, and renal impairment. Half of the patients with aHUS develop end-stage kidney disease (ESKD) [1]. Genetic testing is crucial for the diagnosis of aHUS, as variants in complement regulatory protein gene significantly increase disease risk. Advances in genetic testing and its widespread use have revealed cases of aHUS recurring after kidney transplantation (KT) [2]. Herein, we present a case of recurrent aHUS after deceased-donor KT (DDKT) in a patient with ESKD of uncertain etiology. This study was approved by the Institutional Review Board of The Catholic University of Korea, Seoul St. Mary’s Hospital (No. KC22ZISI0823).
A 45-year-old man with ESKD of uncertain etiology presented with allograft dysfunction 1 month after DDKT. The patient was on triple immunosuppressive therapy, including tacrolimus (trough level, 11.5 ng/mL). On admission, serum creatinine level was 2.7 mg/dL (baseline level, 1.4 mg/dL). Laboratory tests revealed Coombs-negative hemolytic anemia and thrombocytopenia with decreased complement levels. Schistocytes on a peripheral blood smear test enabled a presumptive diagnosis of TMA. However, allograft biopsy (Fig. 1B–D) revealed focally proliferative glomerulonephritis without pathological findings of TMA on light microscopy and bright glomerular C3 staining on immunofluorescence microscopy. Meanwhile, electron microscopy revealed segmentally thickened capillary basement membranes with subendothelial widening, indicating possibly early TMA. A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity was 63.6%, and the Shiga toxin test result was negative. Despite seven sessions of plasma exchange, the patient’s allograft function did not recover, necessitating subsequent hemodialysis (Fig. 1A).
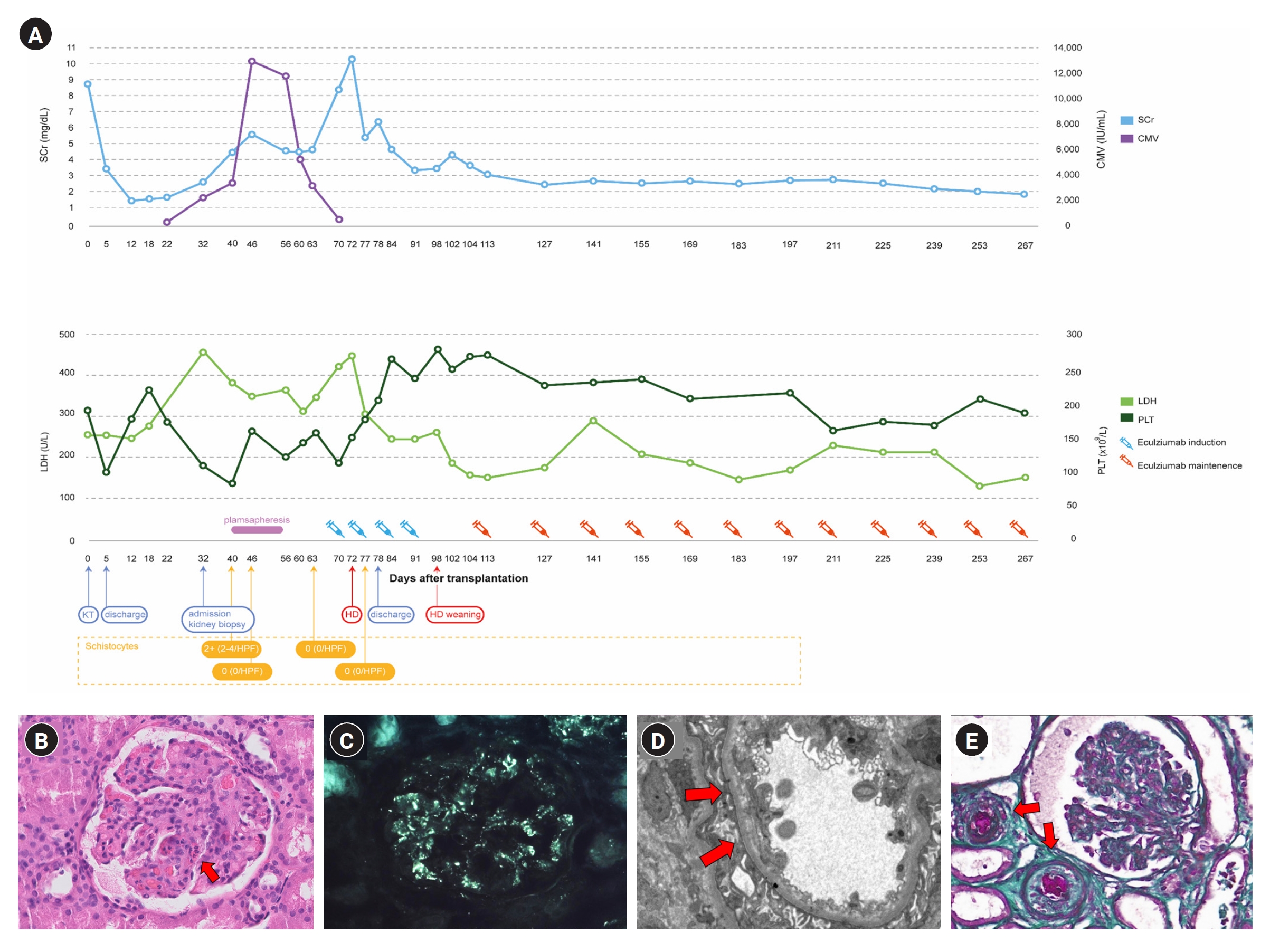
Patient’s clinical timeline and biopsy results.
(A) Timeline of the patient’s clinical course and laboratory findings. (B) Light microscopy on allograft kidney (H&E, ×400). Focally proliferative glomerulonephritis with many neutrophils in capillary loops (arrow). (C) Positive immunofluorescence for C3 showing a diffuse, fine granular pattern of the glomerular basement membrane in allograft kidney. (D) Electron microscopy on allograft kidney. Segmentally thickened capillary basement membranes with endothelial damage and subendothelial widening (arrows). (E) Light microscopy on native kidney (trichrome, ×400). Occluded arteriolar lumen with thrombosis and intimal edema (arrows).
CMV, cytomegalovirus; HD, hemodialysis; HPF, high-power field; KT, kidney transplantation; LDH, lactate dehydrogenase; PLT, platelet; SCr, serum creatinine.
In differentially diagnosing primary aHUS, we performed clinical exome sequencing and discovered a novel heterozygous missense variant (NM_000204.4: c.119A>C, p.His40Pro) of CFI gene (Fig. 2A). Subsequently, the variant was confirmed to originate from the father by direct sequencing of the trio samples (Fig. 2B) and was classified as a variant of uncertain significance (VUS), according to the guideline [3]. Additionally, we employed multiplex ligation-dependent probe amplification (MLPA) to examine copy number variations in CFH and CFH-related genes, and found a pathogenic heterozygous deletion in exon 22 and its downstream region in CFH (Fig. 2C). Furthermore, we found arteriolar thrombotic occlusion upon review of his native kidney biopsy (Fig. 1E). Finally, aHUS was identified as the cause of ESKD and allograft dysfunction. The patient underwent hemodialysis for 4 weeks before completing a 4-week induction with eculizumab (1,200 mg/week). After induction, the patient successfully terminated hemodialysis and, with continued eculizumab therapy (900 mg every 2 weeks), hemolysis resolved and allograft function steadily improved (Fig. 1A).
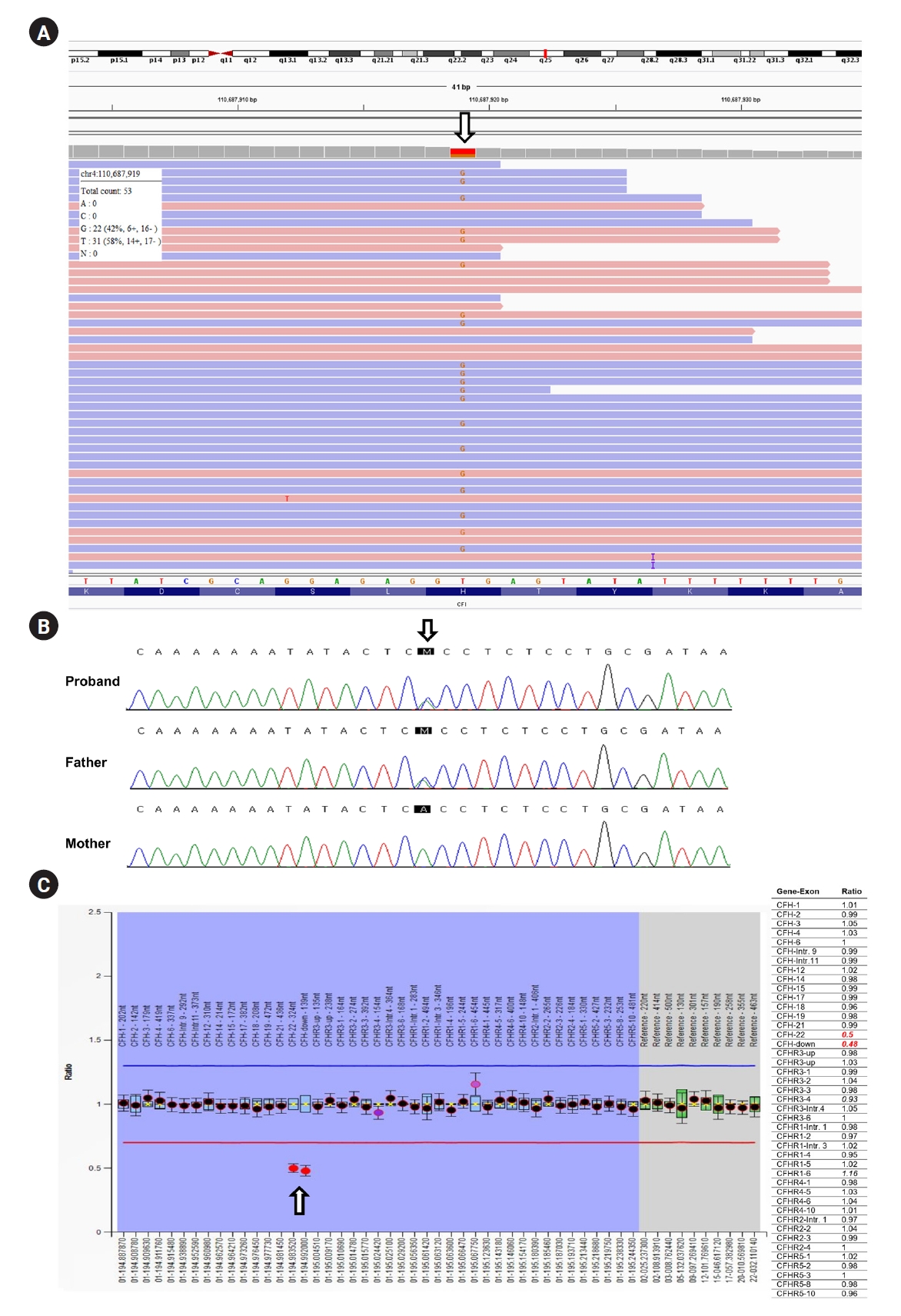
Results of the genetic evaluation.
(A) The clinical exome sequencing result viewed in the Integrative Genome Viewer. A novel heterozygous missense variant, NM_000204.4: c.119A>C, p.His40Pro, is observed in CFI. (B) Results of the direct Sanger sequencing for the patient and his parents. The father is confirmed to carry the same CFI variant. (C) Multiplex ligation-dependent probe amplification shows a heterozygous deletion of exon 22 and its downstream in CFH.
Recent registry data [4] have shown that up to 20% of KT recipients have ESKD of uncertain etiology. Groopman et el. [5] conducted whole-exome sequencing for 3,315 patients with chronic kidney disease and identified diagnostic variants in 307 patients (9.3%). Ten variants were associated with TMA; however, none were clinically diagnosed with TMA before genetic evaluation. This suggests that aHUS may be underdiagnosed or misdiagnosed in patients with ESKD.
The main pathological features of TMA are arteriolar and capillary thrombosis with characteristic abnormalities in the endothelium and vessel wall [1]. However, unlike typical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura, aHUS biopsies rarely show thrombi [6]. Herein, no prominent vascular thrombosis was observed under light microscopy; however, electron microscopy revealed endothelial damage, indicating early-stage disease and the potential for allograft salvage.
We identified a novel missense CFI variant, c.119A>C. However, the patient’s clinical course was more severe than that expected for a CFI VUS carrier. Techniques such as MLPA allow the identification of CFH/CFH-related 1 (CFH/CFHR1) hybrid alleles, which are undetectable by sequencing and present in approximately 3%–5% of all aHUS cases [7]. Using MLPA, we discovered a partial deletion of CFH, predisposing the patient to aHUS. However, the locations of other CFHR deletions could not be identified; therefore, it was difficult to determine which CFH/CFHR hybrid was generated, which requires further research.
Eculizumab, a humanized monoclonal anti-C5 antibody, is a well-documented therapeutic agent for aHUS. Eculizumab prophylaxis significantly reduces recurrence rates and its timely use improves allograft survival in patients with recurrent aHUS [8]. However, the optimal duration for its maintenance remains inconclusive [8]. As the prognosis of aHUS is highly dependent on genetic variants, individualized risk stratification based on the genetic background may be necessary to determine appropriate treatment durations. As our patient harbors a CFH variant belonging to the high-risk variant [8], treatment discontinuation should be approached with utmost caution.
In summary, we report a case of aHUS that was overlooked in a patient carrying a heterozygous deletion of CFH until recurrence occurred post-DDKT. This report emphasizes the importance of primary kidney disease evaluation and careful monitoring for TMA, especially in patients with ESKD of uncertain etiology. Genetic testing is crucial for aHUS diagnosis, regardless of typical TMA features in pathology. Prompt initiation of eculizumab can prevent allograft failure.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Funding
This study was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI22C1529), and by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (RS-2023-00209312).
Data sharing statement
The data presented in this study are available upon reasonable request from the corresponding author.
Authors’ contributions
Conceptualization: Haeun Lee, HSK, Hanbi Lee
Data curation: Haeun Lee, HSK, SHE, IOS, JS
Visualization: Haeun Lee, HSK, JS
Writing-original first draft: Haeun Lee, HSK
Writing-review & editing: YJC, CWY, MK, BHC
All authors read and approved the final manuscript.
Acknowledgements
The authors thank Hayoung Lee for designing Fig. 1A.