The impact of hypoxia-inducible factors in the pathogenesis of kidney diseases: a link through cell metabolism
Article information

Abstract
Kidneys are sensitive to disturbances in oxygen homeostasis. Hypoxia and activation of the hypoxia-inducible factor (HIF) pathway alter the expression of genes involved in the metabolism of renal and immune cells, interfering with their functioning. Whether the transcriptional activity of HIF protects the kidneys or participates in the pathogenesis of renal diseases is unclear. Several studies have indicated that HIF signaling promotes fibrosis in experimental models of kidney disease. Other reports showed a protective effect of HIF activation on kidney inflammation and injury. In addition to the direct effect of HIF on the kidneys, experimental evidence indicates that HIF-mediated metabolic shift activates inflammatory cells, supporting the HIF cascade as a link between lung or gut damage and worsening of renal disease. Although hypoxia and HIF activation are present in several scenarios of renal diseases, further investigations are needed to clarify whether interfering with the HIF pathway is beneficial in different pathological contexts.
Background
The number of patients with end-stage renal disease is increasing, largely due to the aging of the population and increased incidence of hypertension and type 2 diabetes [1,2]. Several hemodynamic [3], inflammatory [4,5], and metabolic [6,7] factors are related to progressive loss of renal function and replacement of the renal parenchyma by fibrotic tissue. Recent studies have demonstrated that changes in the metabolism of podocytes and tubular cells and/or immune cells in kidneys are involved in the pathogenesis and progression of kidney diseases [7–9].
Podocytes require a high energy supply to maintain their cellular functions [10]. Dysfunctions in energy metabolism may result in cell damage and glomerular diseases [9,11]. Dysregulation of podocyte mitochondrial dynamics and function is associated with development of focal segmental glomerulosclerosis (FSGS) [12]. After exposure to high glucose, podocytes show a significant increase in oxygen consumption rate (OCR) and a higher OCR after addition of transforming growth factor beta (TGF-β), indicating prominent changes in their energy metabolism by stimuli related to diabetic kidney disease, and such metabolic effects may sensitize the cells toward apoptosis [13]. Mitochondrial dysfunction also plays a role in the crosstalk among glomerular cells in renal diseases. Activation of TGF-β signaling in podocytes induces endothelin-1 release, which promotes mitochondrial dysfunction in adjacent endothelial cells and contributes to the development of glomerular lesions in experimental FSGS [14].
Glomerular lesions can extend to the tubulointerstitial area with the passage of protein and pro-inflammatory material to the neighboring interstitium and consequent inflammation and fibrosis [15]. However, interstitial fibrosis can start and become chronic in the absence of glomerular lesions. This process involves the participation and interaction of several cell types, including fibroblasts and tubular epithelial cells [16,17]. Recent studies have shown that the fibrogenic process in the kidneys is also associated with alterations in the metabolism of tubular epithelial cells [18,19]. Inhibition of genes related to regulation of the oxidative metabolism of fatty acids in tubular epithelial cells results in ATP depletion, intracellular lipid deposition, cell death, and dedifferentiation, as are observed in renal fibrosi [18]. The “Warburg effect,” which is the cellular metabolic reprogramming that exchanges oxidative metabolism for activation of aerobic glycolysis [20], may also be involved in the damage to tubular epithelial cells in kidney diseases. However, whether such changes in energy metabolism occur in tubular epithelial cells and what metabolic pathways are altered in the process of renal fibrosis remain unclear.
Kidney-resident immune cells participate in the initiation and spread of kidney damage and respond to metabolic shifts by changing their phenotype and functions. Macrophages are plastic cells; their phenotype can polarize to pro-inflammatory or anti-inflammatory, which require different metabolic pathways. Pro-inflammatory macrophages (M1) rely mainly on glycolysis, whereas anti-inflammatory macrophages (M2) have high oxidative phosphorylation (OXPHOS) activity and fatty acid oxidation [7]. Under homeostatic conditions, kidney-resident immune cells exhibit OXPHOS as the predominant metabolic pathway. Affected kidneys are infiltrated by pro-inflammatory cells, and predominance of glycolytic activity is observed [21].
The maintenance of renal cell energy homeostasis is under the control of a series of pathways and molecules involved in modulation of cell metabolism, among which the hypoxia-inducible factor (HIF) pathway plays an important role [22].
Hypoxia-inducible factor: mechanism and function
HIFs are oxygen-sensitive transcription factors that act in metabolic reprogramming to provide cell adaptation under hypoxic conditions [23]. HIF proteins consist of two subunits, α and β, that form a functional complex. The α subunit is unstable, and three distinct HIF-α have been identified: HIF-1α, HIF-2α, and HIF-3α [24,25]. The β subunit is stable and always present in the protein complex. Once the functional complex is formed through dimerization of the two subunits, the complex translocates to the nucleus and activates target genes and cofactors [25]. Studies have shown that HIFs can regulate the expression of genes related to iron regulation, glycolysis, cell survival, erythropoiesis, apoptosis, and angiogenesis [26,27].
HIF-1α and HIF-2α are differently distributed in organs and cells. Both contain a dimerization domain in the N-terminus and a transactivation domain in the C-terminus and regulate common target genes such as facilitated glucose transporter 1 (GLUT1), vascular endothelial growth factor (VEGF), and adrenomedullin genes [25,28,29]. Both HIF-1α and HIF-2α regulate physiological and pathological angiogenesis [30,31] and also regulate specific target genes. HIF-1α is responsible for glycolytic and carbonic anhydrase-9 gene regulation, while HIF-2α regulates TGF-α and cyclin D1 genes [32,33]. In addition, Elvert et al. [34] observed that expression of the VEGF receptor 2 gene is induced by HIF-2α but not by HIF-1α. Less is known about the role of HIF-3α; however, it has been shown to negatively regulate the expression of genes up-regulated by HIF-1α and HIF-2α [35].
HIF activity is regulated by proteasomal-mediated degradation of the HIF-α subunit [36]. Under normoxic circumstances, prolyl hydroxylase domain (PHD) proteins hydroxylate HIF-α at prolyl residues. Once the HIF-α is hydroxylated, the von Hippel–Lindau (VHL) protein E3 ubiquitin ligase ubiquitinates HIF-α for subsequent degradation by the 26S proteasome system [25,37]. The role of PHDs is oxygen dependent; cellular PHD activity is mostly regulated by oxygen partial pressure, but it can also be modulated by reactive oxygen species (ROS), succinate, and nitric oxide (NO) [38,39]. HIF-α can be also acetylated by the arrest defective 1 acetyltransferase, which enhances the interaction between VHL and HIF-α and promotes its degradation [25]. Under hypoxic conditions, HIF-α is modulated by a small ubiquitin-like modifier (SUMO), which enhances the affinity between HIF-α and VHL and promotes PHD-independent degradation [40]. This occurs in the absence of a sentrin/SUMO-specific protease 1 (SENP1). In the presence of SENP1, HIF-α loses the SUMO modification in a process called deSUMOylation, preventing HIF-α degradation. Once HIF-α is stable, HIF-α and HIF-β dimerize, producing a functional transcriptional complex capable of activating hypoxia-response element genes [25,40].
Kidney oxygenation, hypoxia, and hypoxia-inducible factor
Physiological kidney oxygenation involves a balance of O2 demand and supply. Renal O2 consumption is mostly related to solute exchange in tubular cells and renal aerobic glycolysis. Most of the energy required for the kidneys is obtained through aerobic production of adenosine triphosphate (ATP). However, segments of the nephron present in the renal medulla can rely on anaerobic metabolism for energy production, since the vascular architecture promotes a physiologic hypoxic condition in the medullary area [25,41]. The parallel arrangement of peritubular capillaries located along the nephrons allows oxygen to diffuse from the descending branch of the vessel toward the ascending branch, which has lower oxygen tension. This countercurrent exchange of O2 decreases O2 availability in the medulla, even though the kidney receives a high amount of blood perfusion [42]. Due to these specific characteristics, the kidneys are extremely sensitive to stresses that cause hypoxia [43].
Many individual risk factors and environmental and behavioral causes might lead the kidneys to a pathological state of hypoxia. Anemia, hypertension, air pollution, hyperglycemia, smoking, atherosclerosis, and acute kidney injuries (AKIs) are some of the factors that reduce oxygen delivery, promoting renal hypoxia [44]. Once activated by hypoxia, the renal HIF pathway can regulate the gene expression of VEGF, glucose transporters, erythropoietin, and endothelin 1, improving angiogenesis, energy metabolism, and erythropoiesis. Therefore, HIF is crucial for cellular adaptation to hypoxia by improving survival and tissue oxygenation and preventing damaging effects [45–47]. This adaptation involves increase of glycolysis and reduction of cellular oxygen consumption [48]. In hypoxic conditions, HIF upregulates the expression of pyruvate dehydrogenase kinase 1 (PDK1) [49], which inhibits the mitochondrial enzyme pyruvate dehydrogenase, repressing the production of acetyl-CoA from pyruvate and decreasing the mitochondrial oxygen consumption in the tricarboxylic acid cycle. Pyruvate is then redirected to the glycolytic pathway [48]. Thus, the HIF pathway promotes a metabolic shift from OXPHOS to glycolysis, decreasing the cellular necessity of oxygen to produce ATP. Moreover, this metabolic shift also protects the cells by reducing ROS formation [50]. Metabolic reprogramming could lead to a cellular bioenergetic crisis once glycolysis produces less ATP than OXPHOS [48]. However, HIF activation leads to an increase in glucose uptake through upregulation of glucose transporters GLUT1 and GLUT3 [48,51] and increases the expression of enzymes involved in the glycolytic pathway [50,51]. In addition, HIF downregulates the medium-chain acyl-CoA dehydrogenase and the long-chain acyl-CoA dehydrogenase enzymes [52] and inhibits carnitine palmitoyltransferase A1 [53], decreasing the transport of fatty acid in the mitochondria membrane and fatty acid oxidation and further reducing cellular oxygen consumption [48]. When hypoxia is brief and transitory, this mechanism of defense is often effective. However, when hypoxia is prolonged, the defense can be insufficient and pathological cellular pathways are recruited, aggravating the hypoxia state and consuming more oxygen for their processes.
The regulatory activity of HIF-1α and its transcriptional response vary in different renal cell types [54,55]. The energy metabolism of proximal tubular epithelial cells relies mostly on oxidative mitochondrial metabolism. Although the capacity for glycolysis is limited in proximal tubular cells, during oxygen deprivation, the transcriptional activity of HIF-1α is activated and promotes the expression of enzymes involved in glycolytic anaerobic metabolism, which can provide small amounts of ATP to renal cells [54]. In podocytes, hypoxia-induced HIF activation increases the expression of VEGF, which promotes vascular development [56] and improves mitochondrial metabolism of other glomerular cells such as endothelial cells in a paracrine manner [57]. Due to the key role of cell metabolism in the maintenance of cell viability and integrity, disturbance of oxygen homeostasis and HIF activity can impact the pathogenesis and/or progression of several pathological conditions, including kidney diseases [25,58].
Hypoxia-inducible factors in kidney diseases
In addition to regulating cell metabolism in kidneys [25,59,60], HIF-1α plays a role in the process of renal injury in different conditions [61–63]. Several lines of evidence indicate that HIF-1α acts as a profibrotic effector in kidney diseases. Activation of HIF-1α in tubular epithelial cells by hypoxia was shown to promote epithelial-mesenchymal transition in vitro and was associated with tubulointerstitial lesion in mice that underwent unilateral ureteral obstruction (UUO) and patients with chronic kidney disease (CKD) [64]. Deletion of VHL in tubular cells resulted in HIF-1α stabilization in the renal cortex and increased fibrosis in mice that underwent 5/6 nephrectomy. Pharmacological blockade of HIF-1α with YC-1 [65] or deficiency of HIF-2α in renal interstitial cells [66] ameliorated renal interstitial fibrosis in UUO mice. Inhibition of renal HIF-1α by short hairpin RNA attenuated glomerular damage and tubulointerstitial fibrosis induced by angiotensin II infusion [67] or chronic renal ischemia [62] in rats. Glomerular type I collagen accumulation was reduced by HIF-1α knockout in the NEP25 model of FSGS [68]. Selective deletion of HIF-1α in proximal tubular epithelial cells ameliorated renal fibrosis in an aristolochic acid mouse model of kidney disease [69].
Another study showed that global stabilization of Hif-1α and Hif-2α through genetic inactivation of VHL attenuated the renal inflammatory status of UUO mice [70]. Pretreatment with FG-4487, a PHD inhibitor, induced accumulation of HIF-1α and HIF-2α in tubular cells and reduced kidney injury and apoptosis in rats with AKI induced by renal ischemia/reperfusion injury (IRI) [71]. In vitro, treatment with enarodustat (a pan-PHD inhibitor) or small interfering RNA (siRNA) knockdown of PHD2 reduced the levels of ROS and increased the viability of renal proximal tubule cells with ischemia by blockade of OXPHOS or oxygen-glucose deprivation. These protective effects of PHD inhibition were attributed to the HIF-1α–induced expression of enzymes involved in glycogen synthesis, with enhanced glycolysis and delayed ATP depletion [72]. In cisplatin-treated mice, pretreatment with roxadustat (FG-4592), a novel PHD inhibitor, enhanced HIF-1α in tubular cells, improved renal function, and reduced markers of renal inflammation/injury [73]. Roxadustat reduced renal crystal deposition and ameliorated renal dysfunction and tubulointerstitial damage in a model of CKD by adenine overload [74]. However, roxadustat had no effect on renal fibrosis or macrophage infiltration in UUO mice [75]. Oral administration of roxadustat corrected anemia and reduced serum hepcidin level, which was increased by inflammation [76], in dialysis and nondialysis patients with CKD [77–79]. Together, these findings suggest that the role of HIF in kidney damage can vary according to the pathological context and the mechanism applied to modulate its activation. Furthermore, therapeutic approaches may interfere with the activation of HIF in cells other than renal cells, such as immune cells.
Physiological hypoxia-inducible factor activity in immune cells
Hypoxia plays a key role in the bone marrow, providing a specific niche for hematopoietic stem cells, maintaining the self-renewal capacity of these cells, and sustaining their survival [80]. Recent research has demonstrated the importance of HIF expression by immune system components in response to physiological hypoxia or inflammation. Evidence shows that activation of HIFs in innate and adaptive immune cells plays a central context-specific manner role in controlling their functions and cell metabolism (Figure 1) [48]. In neutrophils, for example, HIF-1α expression is associated with a metabolic shift to a glycolytic profile, associated with increased phagocytic capacity and formation of neutrophil extracellular traps (NETs), in addition to promoting survival [81]. Human and mouse neutrophils express HIF-2α, which is up-regulated by lipopolysaccharide (LPS). HIF-2α–depleted inflammatory neutrophils derived from murine bronchoalveolar lavage fluid from LPS-challenged mice expressed lower level of the antioxidant enzyme catalase than wild-type neutrophils and underwent increased apoptosis in response to nitrosative stress [82]. In dendritic cells, activation of HIF-1α has been associated with the capacity to migrate and to activate T cells [83]. Recently, HIF-1α expression was also observed in natural killer cells in the skin and was shown to play an important role in the balance between tissue repair and antibacterial defense [84]. Although its participation in metabolism has not been investigated, the expression of HIF-1α was associated with a greater migratory capacity in eosinophils [85].
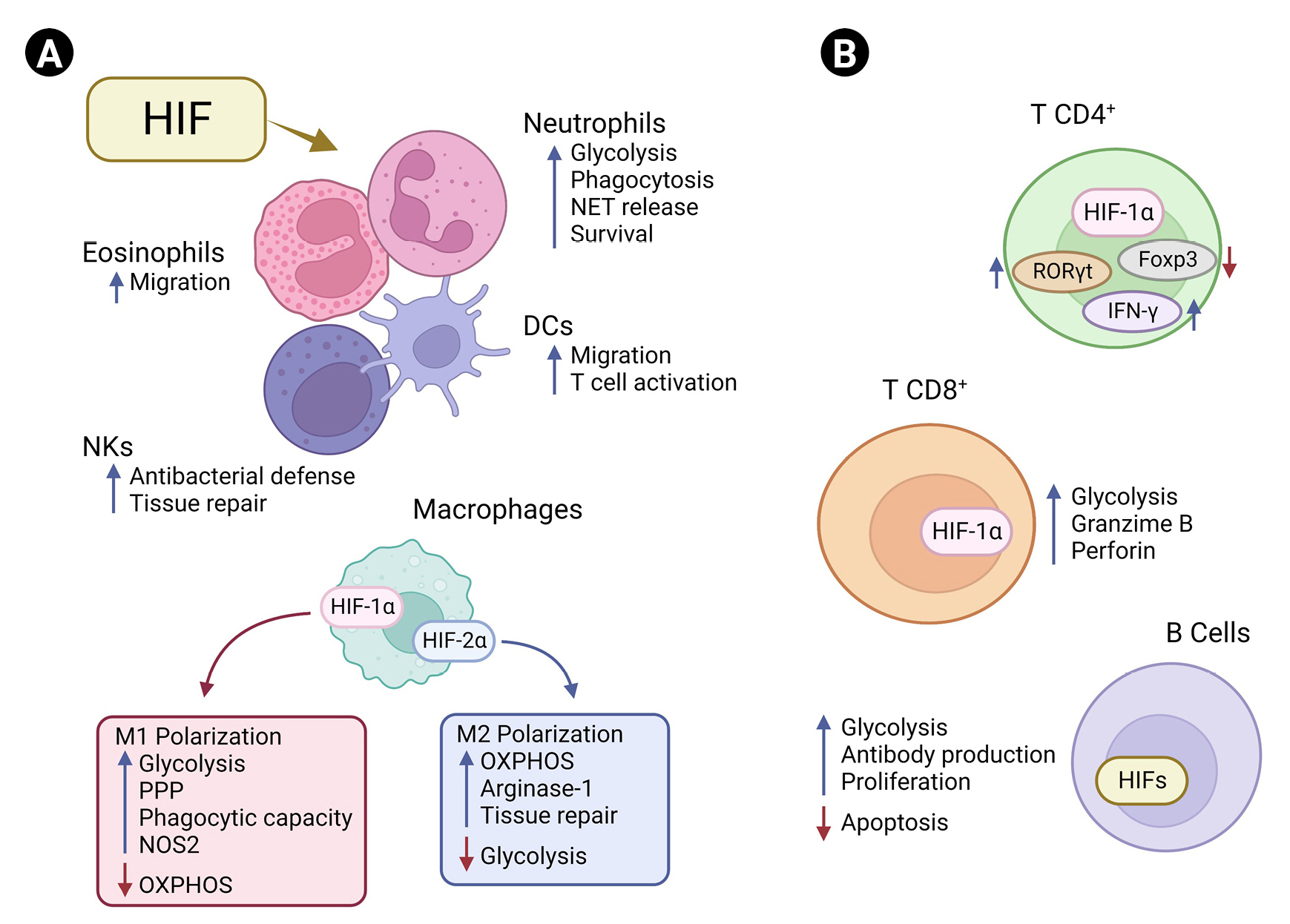
Hypoxia-inducible factors (HIFs) and immune cells.
(A) HIFs regulate immune cells by interfering with their metabolic pathways and function/phenotype. In neutrophils, HIF-1α activation favors glycolysis, increasing the cellular phagocytic capacity, surveillance, and release of neutrophil extracellular traps (NET). In natural killer (NK) cells, HIF-1α is associated with the balance between antibacterial function and tissue repair. HIF-1α increases the migration capacity of both eosinophils and dendritic cells (DCs). In pro-inflammatory macrophage (M1), HIF-1α is fundamental for the activation of glycolysis and pentose phosphate pathways (PPP) and increases the phagocytic capacity and nitric oxide synthase 2 (NOS2) expression, while suppressing oxidative phosphorylation (OXPHOS). HIF-2α is associated with higher tissue repair capacity through arginase-1 expression and lower glycolysis in anti-inflammatory macrophage (M2), which rely on OXPHOS to meet their energy demands. (B) HIF pathways balance the expression of transcriptional factors and effector functions in adaptive immunity. HIF-1α downregulates forkhead box P3 (Foxp3) and upregulates the retinoid orphan receptor gamma t (RORγt) in CD4+ T cells, interfering with their immune functions. During Th1 differentiation, HIF-1α increases the expression of interferon gamma (IFN-γ). In CD8+ T cells, HIF is associated with increased release of granzyme B and perforin and enhanced glycolytic activity. In B cells, HIF induces glycolysis, antibody production, and proliferation and inhibits apoptosis.
In macrophages, activation of the glycolytic pathway through expression of HIF-1α is associated with a pro-inflammatory profile (M1), increasing the phagocytic capacity and favoring the production of cytokines essential for clearance of viruses and bacteria [86,87]. Tannahill et al. [88] demonstrated that LPS-induced HIF-1α stabilization leads to a metabolic shift toward glycolysis and the pentose phosphate pathway (PPP) in macrophages by inducing the expression of genes essential for glycolysis, which results in accumulation of intermediates of the Krebs cycle such as fumarate, malate, and succinate. Increasing succinate in macrophages leads to inhibition of PHDs through generation of ROS by mitochondrial reverse electron transport from complex I or even by competition for the binding site of its cosubstrate, alpha-ketoglutarate [89,90]. This inhibition finally results in accumulation of HIF-1α, which induces a pro-inflammatory phenotype in macrophages, increasing the production of inducible NO synthase, which drives NO synthesis [86,88,91]. Notably, suppression of HIF-2α increases the levels of NO and efferocytosis in macrophages [91,92]. This demonstrates that the HIF-α isoforms regulate NO production and phagocytic capacity in macrophages in an antagonistic way (Figure 1A).
HIF-1α also participates in several processes essential for the development and functioning of T and B cells and generation of antibodies upon an antigenic challenge (Figure 1B) [93,94]. Cho et al. [93] demonstrated that HIF signaling increases the glycolytic metabolism of germinal center B cells, affecting their antibody production, proliferation capacity, and apoptosis. In line with this finding, another group observed that the deletion of VHL leads to a decrease in antigen-specific terminal center B cells, impairing the generation of high-affinity antibodies [94]. Activation and differentiation into subtypes of CD4 T cells and the cytotoxic capacity of CD8 T cells have also been associated with induction of the HIF-1α–dependent glycolytic pathway [81]. HIF-1α promotes the differentiation of Th17 cells through transcriptional activation of the retinoid orphan receptor gamma t (RORγt), the main transcription factor of Th17 cells. HIF-1α binds to p300/RORγt, and this protein complex binds to the interleukin 17 (IL-17) promoter, enhancing IL-17 gene expression and contributing to Th17 function [95]. In addition, signal transducer and activator of transcription 3 (STAT3), which is activated by IL-21, IL-6, and IL-23, can physically interact with the promoter of HIF-1α and increase its expression [96,97]. Concomitantly, HIF-1α can direct the major transcriptional factor of regulatory T cells (Tregs), forkhead box P3 (Foxp3), to proteasomal degradation [95]. Shehade et al. [98] showed that T cell differentiation under hypoxic conditions had a higher number of IL-17–producing cells, whereas the Foxp3+ Treg frequency was significantly decreased. In Th1 differentiation, HIF-1α can act as an enhancer of the glycolytic pathway and interferon gamma (IFN-γ) expression through retroactive activation of STAT3 [81,98]. The higher amount of HIF-1α in Th1 cells leads to an increase in lactate dehydrogenase A, which increases acetyl-CoA accumulation in the cells, promoting histone acetylation and transcription of IFN-γ [99]. HIF signaling is also important for CD8+ T cell function, promoting the expression of crucial transcription, effector, and costimulatory-inhibitory molecules and favoring the clearance of viruses and tumors. Deletion of VHL and increase in HIF-1α and HIF-2α result in higher effector function in CD8+ T cells during chronic infection, culminating in hyperinflammation [100]. These examples demonstrate the complexity behind the roles of HIF-1α in different immune cells, which also depend on the surrounding cellular environment.
Considering that HIF can alter the metabolism of immune cells, interfering with their inflammatory activity, it is conceivable that its activation acts as a regulatory link between events that impairs kidney function/cell metabolism (e.g., hypoxia, LPS, ROS) and the inflammation that is established and progresses in kidney diseases.
Role of hypoxia-inducible factor in immunity in acute kidney injury
The pathophysiology of renal IRI is accompanied by intense inflammation, and researchers have increasingly demonstrated a relationship between HIF-1α activation and immune cells during this process. Increased transcriptional activity of HIF-1α in renal tubular cells has been closely associated with macrophage-dependent inflammation [101]. The nuclear factor kappa B (NF-κB) pathway is highlighted for its central role in promoting inflammation and is activated by inflammatory cytokines, leading to their retroactive production. Li et al. [101] demonstrated that NF-κB binds the HIF-1α promoter, leading to increased HIF-1α expression and consequent protection against tubular injury during AKI.
Using the cisplatin model of AKI and other models of acute and chronic hypoxic kidney injury, Yamaguchi et al. [102] found that the inflammation-related transcription factor CCAAT/enhancer binding protein δ (CEBPD) is a regulator of HIF-1 in the kidney, binding directly to the HIF-1α promoter and potentiating its transcription. In tubular epithelial cells, CEBPD was rapidly induced by inflammatory cytokines produced by macrophages, such as IL-1β, through NF-κB activation, which increases HIF-1α expression during hypoxia and is essential for non-hypoxic induction of HIF-1α.
Folic acid, also known as vitamin B9, is necessary for one-carbon transfer reactions and nucleic acid synthesis; however, it causes toxicity and AKI when administered in high doses [103]. Interestingly, pretreating human monocyte THP-1 cells with folic acid decreased the nuclear accumulation of HIF-1α protein and reduced the expression of IL-1β and tumor necrosis factor alpha (TNF-α), while it increased the level of IL-10. In addition, KC7F2 (an HIF-1α inhibitor) reduced the levels of these hypoxia-induced cytokines, whereas dimethyloxalylglycine (DMOG, a PHD inhibitor) induced their over-expression [104]. Gentamicin nephrotoxicity is also a common cause of drug-induced AKI. Dose-dependent elevation of renal HIF-1α messenger RNA level and increased tubulointerstitial infiltration of ED1+ macrophages have been reported in rats with gentamicin-induced acute injury [105,106]. Treatment with cobalt activates HIF-1α and reduces macrophage infiltration in kidneys exposed to gentamicin [106].
The AKI to CKD transition and development of fibrosis in mice treated with low-dose cisplatin was accompanied by a significant increase in the total macrophage population, with a higher amount of M2 macrophages expressing arginase 1 [107], which is induced by HIF-2α and suppresses NO production [91]. A recent study investigated the role of macrophages in response to repeated low doses of cisplatin-induced fibrosis using liposome-encapsulated clodronate to deplete macrophages in mice. The authors showed that renal depletion of F4/80high and M2 macrophages with decrease in arginase 1 expression attenuates the development of renal fibrosis, suggesting a pathogenic role for kidney-resident M2 macrophages in the progression of fibrosis [108].
Chronic kidney disease-related complications and hypoxia-inducible factor activation
After AKI, an adaptive repair process can restore the integrity of the renal tubules. In contrast, incomplete repair with undifferentiated and atrophic tubules and persistent inflammation can result in renal fibrosis and progression to CKD.
Hypoxia is an early event in the development and progression of experimental diabetic kidney disease, in which inflammation and macrophage polarization play a key role [109]. TGF-β–activated kinase 1 (TAB1) binding protein and TGF-β–activated (TAK1) binding protein form a complex (TAB1/TAK1) that can activate the NF-κB signaling pathway in bone marrow-derived macrophages, activating HIF-1α, increasing glycolytic metabolism, and promoting polarization of these cells toward the M1 pro-inflammatory phenotype [110]. In contrast, myeloid cell-specific activation of HIF suppressed inflammation in UUO mice, whereas specific inactivation of HIF in these cells enhanced inflammation. Furthermore, prolonged exposure to hypoxia suppressed the expression of multiple inflammatory molecules in non-injured kidneys [70]. Thus, hypoxia and/or HIF activation in myeloid cells seem to attenuate the renal inflammation associated with UUO. Consistent with these findings, we observed that exposure to chronic hypoxia reduced renal infiltration by CD68+ macrophages and attenuated renal oxidative stress, innate immunity activation, and injury in rats with 5/6 nephrectomy, while it did not promote renal injury or inflammation in sham-operated rats [111]. HIF-1 and HIF-2 exert different effects on macrophage function in vitro; HIF-1 promotes polarization to the M1 phenotype, while HIF-2 activation induces the M2 anti-inflammatory phenotype [70].
Immunoglobulin A nephropathy (IgAN) is one of the most common types of primary glomerulonephritis. Infiltration of CD68+ and CD206+ macrophages is increased in the kidneys of patients with IgAN [112,113], and the presence of CD68+ macrophages in the tubulointerstitial area was associated with increased renal activation of NF-κB [114], which binds the HIF-1α promoter and enhances its transcription [101]. Indeed, the expression of HIF-1α has been detected in biopsy material from patients with IgAN. Notably, higher HIF-1α expression was associated with lower serum creatinine level, low interstitial fibrosis score, and low glomerular sclerosis in early CKD. However, once fibrosis progresses in the later stages of CKD, lower HIF-1α expression is detected [115]. Thus, renal expression of HIF-1α may be beneficial in early CKD, when active tissue damage is ongoing.
Cardiovascular complications are the leading cause of death in CKD patients [109]. While HIF-1α induces expression of VEGF, endothelin-1, and matrix metalloproteinases in endothelial cells to facilitate angiogenesis, it induces the proliferation of vascular smooth muscle cells in atheroma by upregulation of CD98 and macrophage migration inhibitory factor. HIF-1α also modulates the function of macrophages derived from diseased foam cells, making the cells more inflammatory and apoptotic [116]. Notably, activation of HIF in immune cells plays different roles in different contexts of CKD, especially in macrophages, promoting chronic inflammation or slowing disease progression. The HIF-mediated inflammatory response resulting from CKD directly affects the kidneys but can reach the circulation and affect other organs.
Lung-kidney crosstalk in diseases: a role for hypoxia-inducible factor?
Lungs and kidneys are related in structure and function. Both organs contain an epithelial barrier that regulates the amounts of fluid and solutes that move across two distinct compartments [117–119]. Evidence indicates a close relationship between kidneys and lungs in several diseases [120,121]. Damaged kidneys can interfere with pulmonary disorders by altering the acid-base or fluid balance or through the production of inflammatory mediators [119]. Lungs are highly susceptible to circulating mediators such as TNF-α, IL-1, IL-6, IL-8, NO, and caspase-3 due to their extensive capillary network [118].
Although the lungs are among the most oxygenated organs in the human body [122], HIFs appear to play a critical role in lung function [123]. HIF-1α is involved in lung vascular development via upregulation of angiogenic factors [124,125]. HIF-2α participates in the formation of alveoli and production of surfactant [126]. HIF-3α knockout mice showed impaired lung remodeling at the late embryonic stage and right ventricular enlargement in the adult stage [127].
HIFs also play an important role in pulmonary diseases [128,129]. Acute lung injury (ALI) is an inflammatory disease characterized by pulmonary edema attributed to increased permeability in endothelial cells and infiltration of protein-rich fluid into the alveoli, which reduces the efficacy of air exchange and can result in hypoxemia and alveolar hypoxia. Sepsis, renal IRI, severe traumatic injury, and cigarette smoking are some of the major risk factors for ALI. Although there is no evidence that hypoxia is a direct cause of ALI, studies suggest that hypoxia can contribute to the pathogenesis of the disease [129]. Experimental acute exposure to hypoxia resulted in an ALI-like scenario in rats, with increased infiltration of immune cells and vascular leakage in lungs, suggesting that lung injury is perpetuated by alveolar hypoxia [130,131]. Indeed, alveolar hypoxia in ALI can trigger an inflammatory response with immune cell infiltration, especially macrophages, and increased production of inflammatory molecules, such as intercellular adhesion molecule-1, TNF-α, macrophage inflammatory protein-1β, and monocyte chemoattractant protein-1 (MCP-1), aggravating the injury to the lungs [122,131]. Furthermore, the lungs are among the organs that express the highest levels of HIF-2α in hypoxia [132], and this HIF-2α upregulation increases the activation of nuclear factor of activated T cells c2 and the proliferation of pulmonary fibroblasts [133]. In contrast, other studies have reported that hyperoxygenation increased mortality in experimental models of ALI, whereas hypoxia reduced inflammation in the lungs [134]. Augmentation of HIF-1α downregulated the expression of toll-like receptor 4 and TNF-α in macrophages and decreased inflammatory impairment in ALI rats [135]. In cultured pulmonary alveolar type II cells, TNF-α induced the upregulation of HIF-1α and inhibited vasodilator-stimulated phosphoprotein expression, which plays an important role in the impairment of the alveolar-capillary barrier in ALI [136].
HIF-1α also plays a metabolic role in ALI. Administration of DMOG protected alveolar epithelial cells from neutrophil-LPS–induced ATP decline and cell death in vitro, whereas knockdown of HIF-1α with siRNA or inhibition of glycolysis using media containing 2-deoxy-d-glucose abolished the protective effect of DMOG, suggesting that HIF-1α activation protects alveolar epithelial cells in ALI by enhancing their glycolytic activity. In addition, treatment with DMOG protected the alveolar epithelial barrier, improved arterial oxygenation, and prevented lung ATP decline in mice with LPS-induced lung injury [137]. Woods et al. [138] demonstrated that tissue-resident alveolar macrophages can adapt to hypoxia through HIF-1α activation in ALI. Hypoxia or treatment with FG-4592 stabilized HIF-1α in resident alveolar macrophages, increasing their glycolytic function and survival, and ameliorated lung injury in mice, suggesting that therapies inducing HIF-1α in macrophages may be beneficial in ALI [138]. Repeated injuries in the pulmonary epithelium can cause abnormal wound healing, with recruitment of immune cells and activation of fibroblasts for extracellular matrix protein production, such as fibronectin and collagen, and result in pulmonary remodeling/fibrosis, which hinders air exchange and leads to systemic hypoxemia [128]. Inhibition of the HIF-1α/PDK1 axis in lung fibroblasts attenuated bleomycin-induced pulmonary fibrosis in mice [139]. Activation of HIF-1α by hypoxia polarized activated macrophages to a fibrotic phenotype through increasing adenosine A2B receptor expression and production of profibrotic mediators [120]. These findings suggest an important role of HIF-1α as an amplifier of pulmonary fibrosis.
Patients with chronic obstructive pulmonary disease (COPD) present long-term respiratory symptoms and airflow limitation caused by remodeling of lung structure, leading to hypoxic conditions. HIF-1α has been implicated in the increase of deoxycytidine kinase, which is responsible for accumulation of deoxyATP and apoptosis in COPD [140]. HIF-1α also promotes mucus hypersecretion in COPD by increasing the expression of mucin 5AC, a major component of airway mucus, in airway epithelial cells [141]. CKD has been shown to affect the long-term survival of COPD patients [142]. Gjerde et al. [143] reported that systemic inflammatory markers are associated with a higher risk of renal failure in COPD patients.
Patients with AKI are twice as susceptible to respiratory failure as those without AKI and can progress to ALI [144]. Mice who underwent ALI after induction of AKI had increased lung neutrophilia in bronchoalveolar lavage fluid and elevated MCP-1 levels in kidneys, serum, and lungs compared with ALI mice without AKI [145]. Moreover, after kidney ischemia, lungs present an inflammatory profile, with increased concentrations of pro-inflammatory cytokines and chemokines, upregulation of caspase-dependent apoptosis, and increased macrophage-mediated pulmonary vascular permeability [146]. Pulmonary stabilization of HIF-1α had a protective effect in mice exposed to LPS by inducing the expression of the antioxidant enzyme heme oxygenase-1 (HO-1) [147]. Notably, hemin-induced HO-1 production reduces the systemic inflammation and improves the renal outcomes after IRI. In addition, treatment with hemin ameliorated lung inflammation in AKI mice [148]. These findings suggest the existence of lung-kidney crosstalk in different pathological scenarios and that HIF-induced antioxidant enzymes as well as HIF-1α target genes play a role in this process (Figure 2).
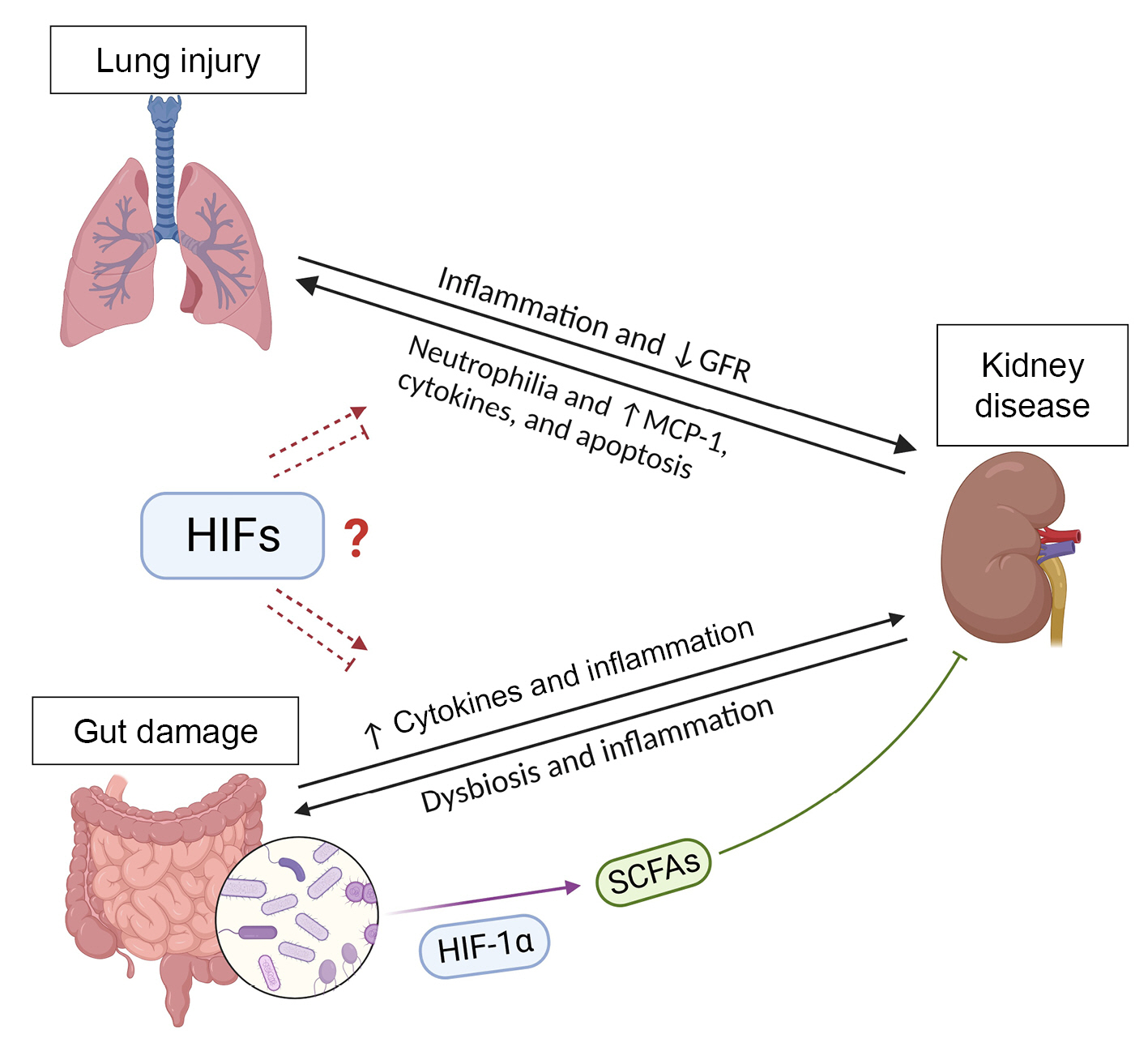
Hypoxia-inducible factors (HIFs) in the lung-kidney and gut-kidney axes.
Increased systemic inflammation is associated with a higher risk of renal failure in patients with chronic obstructive pulmonary disease. Presence of kidney disease increases neutrophilia in bronchoalveolar lavage fluid and the levels of monocyte chemoattractant protein-1 (MCP-1), cytokines, and apoptosis in the lungs. HIF activation induces the expression of the antioxidant enzyme heme oxygenase 1, which can reduce the injury to the damaged lungs and kidneys. Renal damage also results in accumulation of uremic toxins, which promote intestinal dysbiosis and inflammation. Gut dysbiosis and lesions on the intestinal epithelium increase the passage of inflammatory molecules from the gut to circulation, reaching the kidneys, where they promote inflammation and dysfunction. A balanced gut microbiota produces short-chain fatty acids (SCFAs), which are increased by HIF activation and exert positive effects on the intestinal mucosa and immunity and reduce kidney inflammation. Therefore, kidneys, gut, and lungs receive a large blood supply, resulting in high exposure to circulating immune cells and molecules. HIFs can interfere with cell metabolism and the expression of cytokines by immune and epithelial cells, increasing the possibility that HIFs play a role in the lung-kidney and gut-kidney axes. However, it remains unclear whether HIF activation is beneficial or acts as a link between the diseases.
GFR, glomerular filtration rate.
The gut-kidney axis: a role for hypoxia-inducible factor?
Oxygen concentrations along the intestinal tract and large intestine are lower than those in other organs such as the lung, liver, and heart [149]. The partial pressure of oxygen level is low in the gut due to the anatomical juxtaposition of the outermost mucosal surface with the oxygen-depleted lumen and a countercurrent oxygen exchange system in the intestinal villi [149,150]. In addition, the gut contains a diverse and dense microbial population that includes aerobic facultative and anaerobic bacteria necessary for breakdown of food nutrients and regulation of intestinal and systemic immune responses [151,152].
HIFs are crucial for O2 regulation in the gut. In hypoxic conditions, HIFs promote the expression of factors responsible for adaptation of the gut to hypoxia, such as genes involved in erythropoiesis, angiogenesis, and metabolism [39]. In addition to the protective barrier in the gut composed of mucus, intercellular tight junctions, and adherens junctions in the external (luminal) and internal (vascular) compartments, HIF-1α was also shown to participate in protection of the intestinal epithelia during intestinal hypoxia [153,154]. Moreover, HIF-1α and HIF-2α have been shown to modulate intestinal epithelial barrier integrity, function, and homeostasis in colitis [155,156].
HIFs play critical roles in inflammatory response in the intestinal tissue. Acute intestinal inflammation involves the accumulation of neutrophils and can progress to chronic inflammation. Recruitment of neutrophils reduces O2 level in the environment sufficiently to activate HIF-1 in the gut of acute colitis model mice [157]. Intestinal tissue from patients with active ulcerative colitis showed increased positive staining for HIF-1α, whereas tissue from patients with Crohn disease showed intense expression of HIF-2α [158]. HIF-1α and HIF-2α were shown to upregulate the expression of creatine kinases in intestinal epithelial cells, which promotes rapid ATP generation via the phosphocreatine-creatine kinase system, improving cellular bioenergetics and reducing the damage to the intestinal epithelium in colitis [156]. Inhibition of PHD with FG-4497 resulted in stabilization of HIF-1α and reduction of intestinal inflammation in murine colitis [159]. Deficiency of PHD1 ameliorated colitis in mice by reducing apoptosis in the inflamed colon and enhancing epithelial barrier function [160]. Wild-type mice treated with AKB-4924 (a PHD inhibitor) showed reduced serum levels of endotoxin, IL-1β, IL-6, and TNF-α and preservation of the intestinal barrier during colitis, whereas mice with epithelial-specific HIF-1α deficiency had no protection against colitis in the presence of AKB-4924 treatment, suggesting that HIF-1α is essential for the modulation of gut inflammatory diseases [161].
The roles of the gut microbiota in the healthy gut and in intestinal diseases have been extensively investigated. A regulated host-microbiota interaction is essential for physiologic homeostasis and regulation of the immune system [151,162]. Dysbiosis is the imbalance of the gut microbiota with changes in the host metabolic capacity and inflammatory responses that can result in damage to different organs. Lifestyle and environmental factors, use of antibiotics, and a diet poor in fibers and high in sugar and fat are associated with dysbiosis [151,163]. Alterations in the gut microbiota composition with a predominance of pathogenic bacteria lead to the synthesis of harmful molecules that damage the gut and are released to the circulation [151,164,165]. Dysbiosis and lesions on the intestinal epithelium also increase the passage of bacteria and other inflammatory players from the gut to the circulation and therefore to other extraintestinal sites such as the kidneys, where they promote inflammation (Figure 2). Loss of renal function results in accumulation of uremic toxins, which promotes gut dysbiosis. This gut-kidney crosstalk also involves immune cells and cytokines that are regulated by bacterial metabolites [151]. A balanced gut microbiota produces short-chain fatty acids (SCFAs) such as acetate, butyrate, and propionate through anaerobic fermentation of dietary fibers or through metabolism of amino acids [166]. SCFAs play an important role in intestinal homeostasis and exert positive effects on the intestinal mucosa and immune response as well as reduce kidney inflammation [151,166]. Butyrate is one of the main SCFAs that act in gut homeostasis [166]. In the healthy gut, butyrate concentration can exceed 30 mM and is a significant energy source for colonic epithelial cell metabolism. Thus, changes in microbiota can result in abnormal colonocyte function [167]. In colonocytes, absorbed butyrate can be converted to acetyl-CoA through β-oxidation in mitochondria, contributing to the consumption of oxygen and activation of HIF-1α and transcription of its target genes [168]. Zhou et al. [169] demonstrated that intestinal epithelial-specific deletion of HIF-1α changed the composition of the gut microbiota and decreased butyrate production, increasing the susceptibility of mice to induced colitis. In addition, butyrate can upregulate the expression of tight junction proteins in the intestinal epithelium in an HIF-1α-induced manner, which reduces intestinal inflammation and protects the barrier function of the gastrointestinal tract [170,171]. Germ-free and antibiotic-treated mice had reduced colonic butyrate content and weaker HIF activation, both of which were restored by butyrate supplementation [172]. Koury et al. [173] showed that the prolonged increase in HIF-1α after experimental gut IRI is mediated by contact of bacterial products within the gut lumen with the stressed intestinal mucosa. These findings suggest a role of the HIF-1α pathway on the protective effect of SCFAs produced by the gut microbiota in intestinal inflammatory diseases.
Evidence indicates that SCFAs produced by the intestinal microbiota also exert a renoprotective effect in AKI. Acetate, propionate, and butyrate improved renal function and reduced inflammation in experimental IRI-induced AKI mice. Furthermore, treatment with SCFAs reduced activation of NF-κB signaling, production of ROS, and translocation of HIF-1α to the nucleus in tubular epithelial cells stimulated with an inflammatory cocktail [174]. Gut dysbiosis and the consequent release of pro-inflammatory cytokines and chemokines by the intestinal epithelium are also involved in the pathogenesis and progression of CKD [162,164,175,176]. This raises the possibility that therapeutic interventions aimed at interfering with renal HIF-1α in kidney diseases also interfere with intestinal HIF activation and microbiota. Kidneys and gut receive a large blood supply, increasing their exposure to HIF-related molecules released by other damaged organs into the bloodstream.
Conclusions
The role of hypoxia and HIF in kidney diseases has become a topic of interest to nephrologists. However, whether HIF activation is beneficial or participates in the pathogenesis and progression of kidney diseases remains unclear. HIF-1α stabilization has been shown to promote epithelial-mesenchymal transition in vitro and act as a profibrotic effector in experimental CKD. In contrast, other studies have reported that HIF-1α activation ameliorates renal inflammation, apoptosis, and injury. HIF also regulates the metabolism of immune cells and can modulate immune responses in kidney diseases. Furthermore, evidence suggests HIF-1α activation as a mechanism linking lung and gut damage to worsening kidney disease. Further studies on the role of HIF in the kidney-lung crosstalk and in the kidney-gut axis are needed. Current research supports HIF-1α and its transcriptional activity as important therapeutic targets to prevent human kidney diseases. However, variations in the HIF response to different pathological context need to be considered when targeting the HIF pathway.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Funding
This work was supported by the São Paulo Research Foundation (FAPESP, grant No. 2017/05264-7, 2019/02893-9, 2021/06748-3, 2022/03740-4, and 2022/01226-1), the National Council for Scientific and Technological Development (CNPq), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.
Data sharing statement
The data presented in this study are available on request from the corresponding author.
Authors’ contributions
Conceptualization: OFN, NOSC
Writing–original draft: All authors
Writing–review & editing: OFN, NOSC
All authors read and approved the final manuscript.
Acknowledgements
The figures were created with BioRender.com.