Renal intercalated cells and blood pressure regulation
Article information
Abstract
Type B and non-A, non-B intercalated cells are found within the connecting tubule and the cortical collecting duct. Of these cell types, type B intercalated cells are known to mediate Cl− absorption and HCO3− secretion largely through pendrin-dependent Cl−/HCO3− exchange. This exchange is stimulated by angiotensin II administration and is also stimulated in models of metabolic alkalosis, for instance after aldosterone or NaHCO3 administration. In some rodent models, pendrin-mediated HCO3− secretion modulates acid-base balance. However, the role of pendrin in blood pressure regulation is likely of more physiological or clinical significance. Pendrin regulates blood pressure not only by mediating aldosterone-sensitive Cl− absorption, but also by modulating the aldosterone response for epithelial Na+ channel (ENaC)-mediated Na+ absorption. Pendrin regulates ENaC through changes in open channel of probability, channel surface density, and channels subunit total protein abundance. Thus, aldosterone stimulates ENaC activity through both direct and indirect effects, the latter occurring through its stimulation of pendrin expression and function. Therefore, pendrin contributes to the aldosterone pressor response. Pendrin may also modulate blood pressure in part through its action in the adrenal medulla, where it modulates the release of catecholamines, or through an indirect effect on vascular contractile force. This review describes how aldosterone and angiotensin II-induced signaling regulate pendrin and the contributory role of pendrin in distal nephron function and blood pressure.
Introduction
Volume contraction increases the release of angiotensin II and aldosterone, which increases renal NaCl absorption, thereby correcting the volume deficit [1]. During volume overload, this response is down regulated, thereby reducing distal nephron NaCl absorption [1]. Most distal NaCl transport occurs within the distal convoluted tubule (DCT), the connecting tubule (CNT), and to a lesser extent, the cortical collecting duct (CCD) [1]. Because the DCT cannot be perfused in vitro, the magnitude of NaCl absorption in this segment cannot be directly quantified. However, studies that directly measured ion flux in renal tubules perfused in vitro demonstrated that net NaCl absorption in the CNT per mm tubule length is approximately twice the absorption in the CCD [2]. Moreover, since the total length of the CNT in vivo is ~6-fold higher than the CCD [1], cumulative NaCl absorption in vivo is greater in the CNT than in the CCD by at least one order of magnitude. While these more proximal segments mediate greater net NaCl absorption than the medullary collecting duct, this latter segment is nevertheless critical in the final regulation of NaCl balance [1].
Experiments using CCDs and CNTs perfused in vitro have also enabled characterization of transport in the CCD and CNT. However, transport in the CCD is much better understood than transport in the CNT since the latter segment is so technically challenging to perfuse in vitro. The CCD is made up of principal and intercalated cells [1]. However, the CNT is composed of CNT and intercalated cells [1,3]. In the CCD, principal cells mediate most Na+ absorption [1,4,5], whereas most Cl− absorption occurs either by intercalated cell-mediated transcellular transport or through paracellular transport [6].
Pendrin is expressed in the apical domains of type B and non-A, non-B intercalated cells, which represent cell types found in the CNT and the CCD [3]. In type B intercalated cells, pendrin mediates Cl− absorption and HCO3− secretion through electroneutral Na+-independent, Cl−/HCO3− exchange [3,7–9]. Apical plasma membrane pendrin protein abundance is greater in the CNT than in the CCD under both basal and stimulated conditions [1,3,10]. This is because the total length of the CNT in vivo is much greater than the CCD and because of the greater pendrin abundance per cell in the CNT than in the CCD.
Models of metabolic alkalosis, such as that seen with aldosterone or NaHCO3 administration [10–14], stimulate pendrin abundance and thereby increase HCO3− secretion and Cl− absorption in the CCD [15,16]. In these treatment models, pendrin knockout mice develop greater alkalosis [10,11], which indicates an impaired ability of these mutant mice to correct the alkalosis. Therefore, the physiological role of type B intercalated cells, and more specifically pendrin, was initially thought to involve restoration of acid-base balance during metabolic alkalosis. However, as will be described in this review, a more important role has been identified in blood pressure regulation and the pressor response to hormones like aldosterone.
The role of intercalated cell subtypes in acid-base regulation
The H+-ATPase is abundantly expressed within intercalated cells. However, H+-ATPase localization within intercalated cells is heterogeneous, since some intercalated cells show apical localization whereas other cells show basolateral or diffuse localization [17,18]. Intercalated cells are therefore classified as type A, type B or non-A, non-B cells, based on their H+-ATPase subcellular distribution and whether or not they express the basolateral Cl−/HCO3− exchanger, AE1 [17,19–21] (Fig. 1 [22]). However, intercalated cell subtypes are also distinguishable by their ultrastructural characteristics [3,23].

Cell types and transporters in the cortical collecting duct (CCD)
Intercalated and principal cell ion transporter distribution within of the CCD is shown. Na+-dependent Cl−/HCO3− exchanger (NDCBE) mediates Na+ and HCO3− absorption, whereas pendrin mediates HCO3− secretion and Cl− absorption. Through the action of these 2 transporters, Cl− and HCO3− are recycled across the apical membrane. Most likely, Cl− is also absorbed through paracellular transport driven by the lumen-negative transepithelial voltage generated by the epithelial Na+ channel (ENaC).
Modified from the article of Wall and Lazo-Fernandez (Annu Rev Physiol 77:363–378, 2015) [22] with original copyright holder’s permission.
The subcellular distribution of the H+-ATPase is thought to determine whether an intercalated cell secretes H+ or HCO3−. Type A intercalated cells secrete H+ through the H+-ATPase (Fig. 1), which is expressed on the apical plasma membrane and is upregulated in models of metabolic acidosis [24,25]. In the type A intercalated cell, this active H+ secretion occurs in parallel with secretion of Cl−, although the mechanism for Cl− secretion is not well understood [26–29].
Type B intercalated cells secrete HCO3− and absorb NaCl through an apical plasma membrane, Na+-independent, electroneutral, Cl−/HCO3− exchanger (pendrin), which is encoded by Slc26a4 [7,9,30,31], and an apical Na+-dependent Cl−/HCO3− exchanger (NDCBE), which is encoded by Slc4a8 [32]. These apical exchangers act serially with net H+ efflux across the basolateral plasma membrane mediated by the H+-ATPase [19–21,31,33,34]. In type B intercalated cells, the basolateral plasma membrane H+-ATPase provides the active step, which generates the driving force for apical anion exchange [35]. In models of metabolic alkalosis, the apical Cl−/HCO3− exchanger and the basolateral membrane H+-ATPase are stimulated, which increases HCO3− secretion by type B intercalated cells [15,36]. From this observation, exchanger-mediated HCO3− secretion is thought to attenuate the alkalosis generated in these treatment models [7,37].
Apical Na+-independent Cl−/HCO3− exchange is also observed in non-A, non-B intercalated cells [38]. Unlike type B intercalated cells, however, the H+-ATPase localizes to the apical, rather than the basolateral, plasma membrane in this cell type [39]. Because this cell type is found primarily in the CNT of the mouse kidney [3], which represents a segment in mouse that has not been successfully perfused in vitro, CNT transport properties, and hence the properties of non-A, non-B intercalated cells, are less well understood than those of type B intercalated cells in the CCD. Nevertheless, given the distribution of Cl− and H+/OH− transporters within non-A, non-B intercalated cells, this cell type most likely absorbs Cl− [40] coupled with the secretion of H+ and HCO3− [38]. Therefore, the luminal fluid of the CNT should ultimately gain CO2 and H2O and lose Cl−. Tsuruoka and Schwartz [38] have shown that CNTs from untreated rabbits secrete HCO3−. If non-A, non-B intercalated cells are the predominant non-type A intercalated cell in this segment, these data suggest that non-A, non-B intercalated cells secrete relatively more HCO3− than H+s under most physiological conditions.
Whether the H+-ATPase of the non-A, non-B intercalated cell provides the active step for apical Cl−/HCO3− exchange, as was observed in the type B intercalated cell [35], is not known. Moreover, in this cell type the relative magnitude of anion exchange-mediated HCO3− secretion and H+-ATPase-mediated H+ secretion is unclear. As such, whether non-A, non-B intercalated cells are acid- or base-secreting cells (or both) is all unclear.
Intercalated cells are thought to regulate renal net acid excretion by secreting H+ or HCO3−. However, as described below, more recent studies have shown that intercalated cells play an important role in fluid and electrolyte balance and blood pressure regulation [9,10].
Pendrin localizes to type B and non-A, non-B intercalated cells in the CCD where it mediates Cl−/HCO3− exchange
In heterologous expression systems, pendrin mediates electroneutral, Na+-independent, Cl−/Cl−, Cl−/HCO3−, and Cl−/I− exchange [41–44]. Pendrin is expressed in the apical regions in a minority of cell types, i.e., type B and non-A, non-B intercalated cells, and colocalizes with NDCBE, which is encoded by Slc4a8 [32]. Because pendrin is an electroneutral Cl−/HCO3− exchanger and because it localizes to the apical region of cells that secrete HCO3− and absorb Cl− through electroneutral Na+-independent, Cl−/HCO3− exchange [7,9,43], we hypothesized that pendrin-mediates this electroneutral anion exchange observed in the rodent CCD. To test this hypothesis, we examined Cl− and total CO2 (HCO3−) transport in CCDs from wild type and pendrin null mice that were administered NaHCO3 and an aldosterone analogue in order to upregulate intercalated cell apical Cl−/HCO3− exchange [7,9,10,15,31]. Because pendrin gene ablation eliminated HCO3− secretion and Cl− absorption observed in CCDs from wild type mice, the apical Cl−/HCO3− exchange observed in the type B intercalated cell is pendrin-dependent [7,9].
Na+ and Cl− exit across the basolateral plasma membrane of type B intercalated cells through Na+-HCO3− cotransporter, AE4 [35], and ClC-K2/barttin Cl− channels [45,46], respectively. H+ exits the cell across the basolateral plasma membrane through the H+-ATPase [34,35].
Cl− absorption in the CCD occurs through 2 separate pathways, which differ in diuretic sensitivity. The first of these is eliminated with the application of epithelial Na+ channel (ENaC) inhibitors, such as amiloride, while the second is blocked with thiazides, which are inhibitors of the NaCl cotransporter (NCC) encoded by Slc12a3 [47]. The magnitude of the amiloride- and the thiazide-sensitive components of NaCl absorption varied between studies and likely depended on the species tested and the treatment model used [32,47]. While thiazides strongly inhibit NCC, NCC cannot mediate thiazide-sensitive Cl− absorption in the CCD because this transporter is not expressed in this segment [32]. However, Leviel et al [32] showed that the thiazide-sensitive component of Cl− absorption observed in the mouse CCD occurs instead through a NDCBE encoded by Slc4a8. The authors also showed that pendrin-mediated Cl−/HCO3− exchange and NDCBE-mediated Na+-dependent Cl−/HCO3− exchange act in parallel to mediate electroneutral NaCl absorption in the CCD. However, in the mouse CCD, the role of NDCBE in Cl− absorption remains controversial [48].
ENaC represents the major mechanism of Na+ absorption by principal cells [4,5]. While ENaC does not directly mediate Cl− transport, ENaC inhibitors markedly reduce Cl− absorption in the CCD [48]. ENaC blockade changes Cl− transport or channel activity either through paracrine signaling or by altering the driving force of Cl− movement. While the mechanism by which ENaC inhibition changes Cl− transport is not known, recent studies have shown that it does not involve pendrin, ClC-5, or CFTR [5,26,48]. Amiloride-sensitive Cl− absorption may involve paracellular Cl− transport or transcellular transport mediated by an electrogenic Cl− exchanger. ENaC-mediated Na+ absorption generates a lumen-negative transepithelial voltage that augments the driving force for electrogenic, transcellular Cl− transport and paracellular Cl− absorption across tight junctions.
Claudin-mediated paracellular Cl− transport is important in both blood pressure regulation and the maintenance of vascular volume. For example, natriuresis, chloriuresis, and reduced blood pressure are observed following claudin 4 gene ablation, particularly when dietary NaCl intake is restricted [49]. Since claudin 4 acts as a Cl− channel and localizes to tight junctions between intercalated cells and principal cells [49,50], ENaC blockade may alter electrogenic Cl− transport in the CCD by eliminating the driving force for claudin 4-mediated paracellular Cl− transport.
The contribution of transcellular Cl− transport relative to paracellular Cl− absorption, and the possible contrasting physiological roles of each, are not well understood. Pei et al [51] had shown that under conditions where energy conservation is needed, the kidney is more dependent on paracellular than on transcellular Cl− absorption. While this study looked primarily at proximal tubule paracellular Cl− absorption, one could speculate that in the CCD, paracellular transport-mediated Cl− absorption is most significant during energy depletion. Since paracellular Cl− absorption is driven by the lumen-negative voltage generated through ENaC-dependent Na+ absorption, ENaC may also be of importance during energy depletion. Conversely, transcellular Cl− transport mediated by type B intercalated cell apical Cl−/HCO3− exchange may be most important when cellular energy is abundant. However, the relative role of each pathway may depend on the need for K+ conservation. For example, in CCDs from mice fed a NaCl-deficient diet, significant thiazide-sensitive NaCl absorption was observed, which reflected NDCBE/pendrin-mediated NaCl absorption [32]. In contrast, in CCDs from mice fed a NaCl-replete diet along with aldosterone, Cl− absorption was unaffected by thiazides, but nearly obliterated with the application of an ENaC inhibitor (benzamil) [48]. Because thiazide-sensitive, NDCBE/pendrin-mediated NaCl absorption is not accompanied by a change in K+ flux [32], whereas ENaC-mediated, amiloride-sensitive NaCl absorption greatly stimulates K+ secretion [52], the thiazide-sensitive, NDCBE/pendrin-mediated component of the mouse Cl− absorption CCD may be most important when K+ conservation is needed.
Pendrin is upregulated with aldosterone
Since aldosterone increases apical anion exchange in the CCD [16,31], other studies have investigated whether aldosterone changes renal pendrin abundance or subcellular distribution. Although the abundance of pendrin at the apical plasma membrane in type B intercalated cells was low under basal conditions [3,10], it increased ~6-fold in response to aldosterone, primarily through sub-cellular redistribution [10]. Moreover, the aldosterone-induced increase in apical anion exchange [15,16,31,53] was eliminated with pendrin gene ablation [7,9]. Therefore, we conclude that both pendrin and type B intercalated cell apical anion exchange are greatly stimulated by aldosterone and that the apical Cl−/HCO3− exchange in type B intercalated cells is mainly pendrin-mediated.
Because pendrin mediates HCO3− secretion in mouse CCD, further experiments have explored the role of pendrin in acid-base balance. As expected, pendrin gene ablation exacerbated the metabolic alkalosis observed with the administration of aldosterone analogues [10]. However, these studies provided surprising evidence that pendrin also modulates blood pressure [10].
Pendrin gene ablation reduces, while pendrin overexpression increases, rodent blood pressure
Since pendrin mediates renal Cl− absorption, we explored its role in NaCl balance [9–11]. Following a NaCl-replete diet, apparent vascular volume was similar in wild type and in pendrin null mice [9,11]. However, following dietary NaCl restriction, pendrin gene ablation increased NaCl excretion, reduced blood pressure, and increased the apparent vascular volume contraction [9,11]. Therefore, the pendrin null kidney has an impaired ability to fully conserve NaCl, which leads to lower blood pressure [9,11,54]. During dietary NaCl restriction, or following an aldosterone infusion, which both upregulate pendrin, the difference in blood pressure between pendrin null and wild type mice was enhanced [9–11,55]. However, since intercalated cells represent only ~1% of total kidney volume [1,56], it is remarkable that this minority cell type had such a significant impact on blood pressure and vascular volume.
Whereas blood pressure was reduced in pendrin null mice, mice that overexpressed pendrin developed hypertension that was very sensitive to dietary Cl− intake [57]. However, whether pendrin-mediated Cl− absorption contributes to NaCl-sensitive hypertension in people has been a topic of great interest.
Pendrin regulates blood pressure and NaCl balance in humans
In 1896, Dr. Vaughan Pendred reported a single family in which congenital deafness and goiter were observed in 2 of the family’s 10 children [58]. This autosomal recessive disorder, called Pendred syndrome, is seen in 7.5 per 100,000 persons [59]. Given the important role it plays in hearing and thyroid function, it was not surprising that the product of the gene responsible for Pendred syndrome (pendrin) is highly expressed in the thyroid and inner ear [60,61]. Early studies, however, had not identified any abnormalities in renal function or blood pressure in people with Pendred syndrome [58]. This view changed, however, after Everett et al [59] had identified and cloned the gene responsible for this syndrome. This discovery facilitated the development of a number of tools, such as anti-pendrin antibodies and pendrin null mice. These tools enabled better study of Pendred syndrome and led to our observations, and the observations of others, showing that pendrin is highly expressed in intercalated cells and that pendrin modulates blood pressure.
After our report showing that Slc26a4 regulates blood pressure in mice [10,55], there was great interest in determining if pendrin also regulates human blood pressure. In the first of these human studies, a retrospective chart review examined the incidence of hypertension in persons with Pendred syndrome and in their unaffected family members [62]. While the study was not adequately powered, it raised the possibility that the absence of pendrin is protective against hypertension. A later study directly investigated blood pressure and NaCl excretion in people with biallelic, inactivating mutations in SLC26a4 [63]. This report demonstrated that pendrin gene ablation reduced blood pressure and induced natriuresis and chloriuresis in humans, similar to what we had shown in our earlier studies in mice.
Pendrin enhances the aldosterone-induced increase in ENaC abundance and function
Although pendrin does not directly mediate Na+ transport, NaCl-restricted pendrin null mice excreted more NaCl than their wild type NaCl-restricted littermates [54]. Therefore, we had asked if pendrin changed the expression of a major renal Na+ transporter [54]. In mice consuming a NaCl-replete diet, where circulating plasma renin and aldosterone were suppressed, pendrin gene ablation did not significantly alter the abundance of renal Na+ transporters such as NHE3, NKCC2, α1Na,K-ATPase, ENaC, and NCC [54]. However, following an aldosterone infusion or dietary NaCl restriction, pendrin gene ablation reduced the abundance of the α, β, and γ ENaC subunits [5,54,55].
Other experiments explored the effect of pendrin gene ablation on ENaC function. Aldosterone administration markedly increased Na+ absorption in the rat and mouse CCD, which was virtually eliminated with the application of ENaC inhibitors [4,5]. Therefore, aldosterone-stimulated Na+ absorption is primarily ENaC-mediated. However, the benzamil-sensitive component of Na+ absorption was reduced by ~60% in CCDs from aldosterone-treated pendrin null mice [5]. These data showed that pendrin gene ablation reduced ENaC abundance and function, which likely contributed to the lower blood pressure observed in these mutant mice.
Aldosterone increases ENaC activity (NPo) by increasing the frequency at which the channel is open (open probability, Po) [64], by increasing the total protein abundance of the ENaC subunits [65], and by increasing the apical plasma membrane ENaC subunit abundance through subcellular redistribution [65]. However, assembly of all 3 ENaC subunits is needed for aldosterone to efficiently deliver ENaC to the apical plasma membrane [66–68].
In other experiments, we performed single channel recordings in principal cells from split open CCDs of aldosterone-treated mice to examine the effect of pendrin gene ablation on channel activity, NPo, channel surface density, N, and channel open probability, Po [5]. Pendrin gene ablation lowered channel activity from the reduced channel surface density due to a decrease in ENaC subunit total protein abundance and ENaC subunit subcellular redistribution [5,54,55]. Pendrin gene ablation also lowered channel activity by reducing channel open probability [5].
Pendrin modulates ENaC abundance in kidney through paracrine signaling
We and others have explored how pendrin changes ENaC abundance and function [55,69]. Because pendrin and ENaC localize to different cell types [3,7,8,70] (Fig. 1), these proteins cannot directly associate. Moreover, pendrin did not change ENaC by altering the level of hormones that regulate ENaC, such as corticosterone, vasopressin, renin, aldosterone, or thyroid hormone [54,71]. Finally, while pendrin gene ablation downregulated ENaC in kidney, pendrin gene ablation did not change ENaC abundance in the thyroid or colon [54]. Moreover, pendrin gene ablation paradoxically increased ENaC abundance in the inner ear [72]. Therefore, the decrease in ENaC subunit abundance and function observed in pendrin null mice appears restricted to the kidney.
Because ENaC function changes with pH [55], we hypothesized that pendrin-mediated HCO3− secretion increases luminal HCO3− concentration, thereby stimulating ENaC. To test this hypothesis, we employed two separate treatment models, each of which increases luminal HCO3− concentration within the CNT and CCD in vivo. In the first, we gave mice NaHCO3 and aldosterone to stimulate pendrin-mediated HCO3− secretion [15,55], thereby increasing luminal HCO3− concentration in the CNT and CCD. In the second, we added a carbonic anhydrase inhibitor (acetazolamide) to the treatment protocol to increase distal delivery of HCO3− from upstream segments, while down regulating pendrin expression and function [55,73]. When mice were treated with aldosterone and NaHCO3, pendrin gene ablation magnified the metabolic alkalosis and reduced renal ENaC subunit abundance and function [55]. However, adding acetazolamide to the treatment protocol eliminated the differences in renal ENaC subunit abundance and function as well as differences in acid-base balance [55]. One explanation for these findings is that with stimulation of distal HCO3− delivery from upstream segments, pendrin null mice are rescued from the enhanced alkalosis and the subsequent decrease in renal ENaC abundance and function.
In other experiments, mouse principal cells (mpkCCD) were used to determine if changing HCO3− concentration in vitro on the apical side of the cell modulated ENaC abundance and function [55]. When HCO3− concentration on the apical side of the monolayer was increased over a period of 3 to 24 hours, ENaC abundance and function increased, which did not depend on the substituting anion. Therefore, pendrin modulates ENaC, at least in part, either through changes in luminal HCO3− or luminal pH.
Since H+-ATPase gene ablation in the kidney reduced renal cortical ENaC abundance and function [69], and since pendrin gene ablation significantly reduced H+-ATPase abundance in type B intercalated cells [74], pendrin gene ablation may reduce ENaC function due to reduced type B intercalated cell H+-ATPase function. Because B1-H+-ATPase gene ablation increased urinary excretion of prostaglandin E2 (PGE2), Gueutin et al [69] had hypothesized that B cell H+-ATPase gene ablation reduced ENaC abundance and function through a PGE2-mediated mechanism (Fig. 2 [22]). They observed that a reduction in intercalated cell H+-ATPase abundance, as occurs with pendrin gene ablation, stimulated ATP secretion. Luminal ATP then acted through purinergic receptors on the apical plasma membrane of principal cells in order to stimulate principal cell Ca2+ release, which increased the production of PGE2, and thereby reduced ENaC abundance and function [69] (Fig. 2). These studies are consistent with a large body of work demonstrating that luminal ATP acts through principal cell apical plasma membrane purinergic receptors in order to reduce phosphatidylinositol 4,5-bisphosphate (PIP2) through a phospholipase C-dependent pathway, which lowers ENaC subunit abundance and function [75–77].
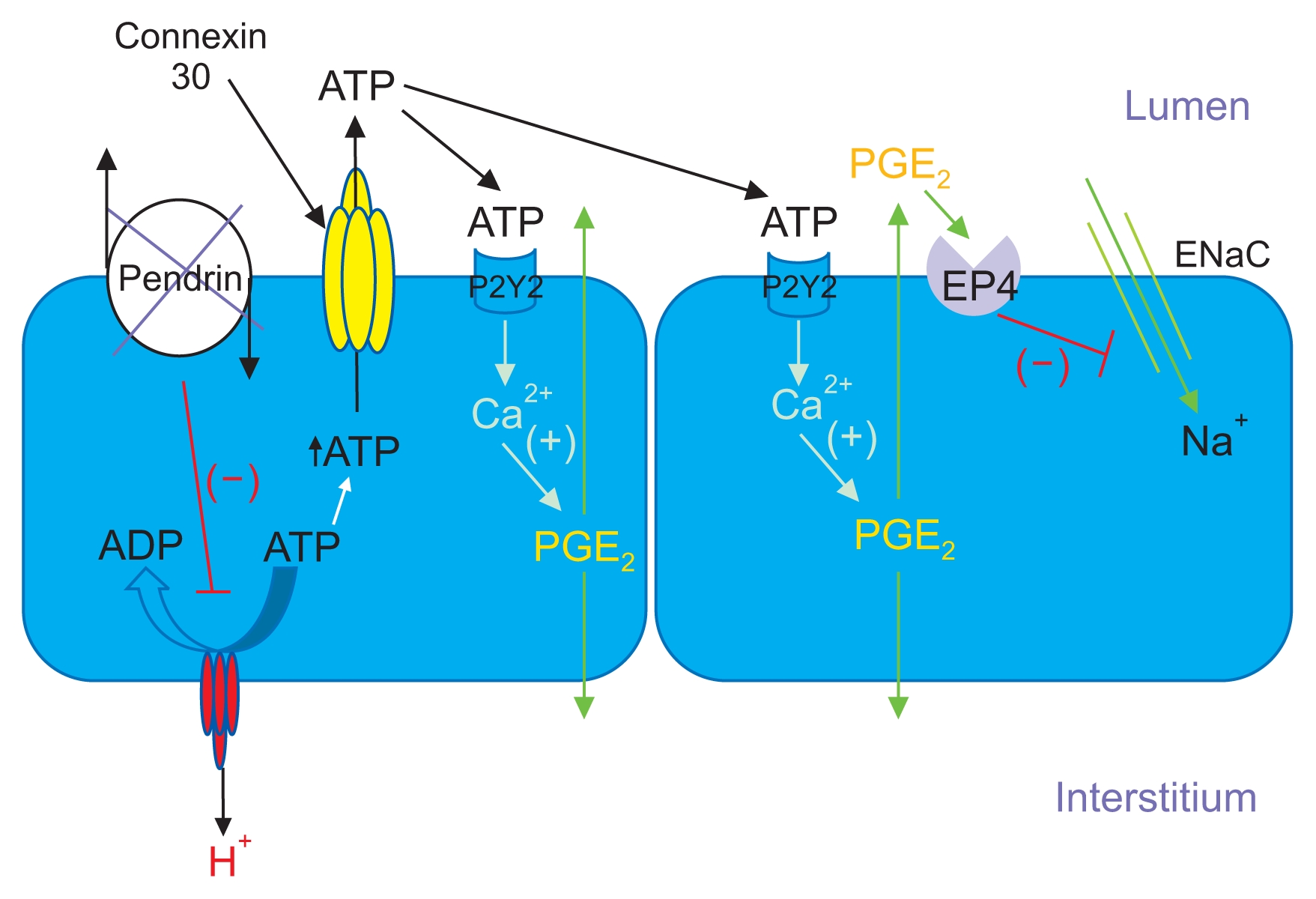
Pendrin gene ablation modulates luminal ATP concentration, which changes ENaC abundance and function
With pendrin gene ablation, H+-ATPase abundance decreases in the type B intercalated cell, thereby increasing intercalated cell ATP content. Luminal ATP concentration then rises through enhanced connexin 30-mediated ATP secretion. Luminal ATP acts on apical membrane purinergic receptors to stimulate calcium release, which increases prostaglandin E2 production (PGE2). PGE2 acts through a receptor-mediated process to reduce ENaC abundance and function.
Modified from the article of Wall and Lazo-Fernandez (Annu Rev Physiol 77:363–378, 2015) [22] with original copyright holder’s permission.
In summary, pendrin gene ablation reduces ENaC abundance and function by changing luminal ATP and HCO3− concentration. However, other pathways may also contribute to this pendrin-ENaC interaction.
Pendrin is upregulated by angiotensin II
Angiotensin II stimulates ENaC-mediated renal Na+ absorption through both short- and long-term mechanisms [78–80]. Therefore, we explored the effect of this hormone on intercalated cell function. In particular, we had asked if angiotensin II application in vitro increased Cl− absorption in the CCD and explored the mechanism by which it occurs [81]. We found that angiotensin II application to the bath induced a 2-fold increase in electroneutral Cl− absorption in CCDs from furosemide-treated wild type mice, but not from pendrin knockout mice [81]. These data indicated that this peptide hormone more likely increased Cl− absorption through transcellular rather than through paracellular transport [81].
Angiotensin II may increase CCD Cl− absorption through a direct effect on apical plasma membrane pendrin abundance or it may stimulate another transporter, thereby increasing the driving force for apical anion exchange. For example, angiotensin II may increase basolateral plasma membrane H+-ATPase abundance in type B intercalated cells, which provides the active transport step for apical anion exchange [35]. To discriminate between these possibilities, we perfused mouse CCDs in vitro in the presence or absence of angiotensin II and then fixed and labeled the tubules for pendrin and the H+-ATPase. Transporter subcellular distribution was quantified by immunogold cytochemistry with morphometric analysis. In type B intercalated cells, we observed no increase in plasma membrane pendrin or H+-ATPase abundance with angiotensin II application in vitro [82] However, angiotensin II induced H+-ATPase subcellular redistribution in type A intercalated cells, which produced a 3-fold increase in its abundance on the apical plasma membrane relative to the cytoplasm [82]. Moreover, this peptide hormone increased HCO3− absorption in the CCD, indicating increased H+ secretion [82], similar to previous observations in the OMCD [83].
In CCDs perfused in vitro, angiotensin II application increased ENaC-mediated Na+ absorption [80] which should have increased the lumen-negative voltage. However, we observed that angiotensin II increased Cl− absorption without changing VT. As such, movement of another ion must shunt the ENaC-mediated current. In the CCD, the lumen-negative VT talls with increased H+ secretion [27], but rises as ENaC-mediated Na+ absorption increases [5]. Since angiotensin II application in vitro increases both H+ secretion and Na+ absorption in the mouse CCD, movement of these 2 ions must shunt the voltage generated by each other.
These data indicate that angiotensin II does not increase pendrin-dependent Cl− absorption in vitro through pendrin subcellular redistribution. Angiotensin II also does not change the driving force for anion exchange by increasing basolateral plasma membrane H+-ATPase abundance. Instead, angiotensin II may increase the driving force for apical anion exchange by lowering intra-cellular Cl− concentration within the type B intercalated cell, such as through activation of a basolateral plasma membrane Cl− channel. However, other mechanisms are possible.
Angiotensin II acts on the angiotensin type 1 receptor to increase apical plasma membrane pendrin abundance through subcellular redistribution, independently of aldosterone [84]. Angiotensin II application in vivo also stimulates the angiotensin type 2 receptor, which acts through nitric oxide to reduce pendrin protein abundance without changing pendrin subcellular distribution [84]. Therefore, the angiotensin type 2 receptor provides a feedback loop, which attenuates the increase in apical plasma membrane pendrin abundance that follows angiotensin type 1a receptor activation by angiotensin II.
Regulation of pendrin by the mineralocorticoid receptor and receptor interaction with aldosterone and angiotensin II
Upon aldosterone binding to the mineralocorticoid receptor (MR), a series of downstream signaling events are triggered, which lead to increased collecting duct transporter or channel activity. In the kidney, the signaling cascade by which aldosterone activates the MR and thereby stimulates ion transport, is best understood for ENaC in principal cells. In addition to aldosterone, cortisol and corticosterone are also MR substrates. However, if a cell expresses 11β-hydroxysteroid dehydrogenase type II, these glucocorticoids are oxidized and inactivated, making aldosterone the preferred receptor ligand [85]. The MR and 11β-hydroxysteroid dehydrogenase type II are clearly expressed in principal cells [85–87]. Convincing MR expression data was also found within type B and non-A, non-B intercalated cells [86,87]. However, because 11β-hydroxysteroid hydrogenase type II expression was either low or absent in intercalated cells [86,88], whether aldosterone is an MR ligand in this cell type is controversial. Aldosterone may activate intercalated cell transporters through the MR, but independently of direct aldosterone binding, such as through oxidative stress or cyclic adenosine monophosphate (cAMP) [89].
Intercalated cell pendrin abundance is reduced with chemical inhibitors of the MR, such as spironolactone [87], either through a direct effect of MR inhibition or through an indirect effect of MR blockade, such as through MR-dependent changes in serum potassium [90]. The reported effect of serum K+ and K+ balance on pendrin abundance has varied between studies, however [12,91–93].
Shibata et al [87] had reported that mouse intercalated cells express a unique MR phosphorylation site. Phosphorylation of the intercalated cell MR at S843 prevented aldosterone binding to the MR, thereby blocking MR activation. However, with increased angiotensin II production, as observed with dietary NaCl restriction, the intercalated cell MR was dephosphorylated at S843 and thereby activated, which augmented aldosterone binding and increased pendrin and H+-ATPase abundance. Because aldosterone-induced S843 MR dephosphorylation was greater with angiotensin II, but reduced with hyperkalemia [87], angiotensin II augments, whereas hyperkalemia attenuates, aldosterone-induced intercalated cell MR activation.
Does pendrin regulate blood pressure through a mechanism that is outside of the kidney?
Blood pressure is regulated not only by the kidney, but also by the central nervous system, by vascular tissue, and through the production of hormones released by the adrenal gland [94]. While studies to date have not demonstrated pendrin expression within the central nervous system, we had observed that pendrin was expressed in both epinephrine- and norepinephrine-producing chromaffin cells within the rat and mouse adrenal medulla where it modulates catecholamine release [95]. While plasma epinephrine and norepinephrine concentrations were similar in wild type and pendrin null mice under basal, unstressed conditions, catecholamine concentrations were higher in pendrin null than in wild type mice after 20 minutes of immobilization stress. Because catecholamine release changes blood pressure, we measured blood pressure before, during, and after immobilization stress, a treatment model associated with increased release of catecholamines. Blood pressure increased in both groups following immobilization stress. However, while blood pressure remained lower in the pendrin null than in the wild type mice before and during the period of stress [95], blood pressure was similar between wild type and pendrin null mice 30 minutes after relief of stress [95]. Therefore, pendrin gene ablation increases catecholamine release, which may limit the drop in blood pressure observed in the absence of renal pendrin-mediated Cl− absorption.
Since blood pressure is the product of systemic vascular resistance and cardiac output, we had explored the effect of pendrin gene ablation on vascular tone [96]. Although we did not observe pendrin expression in vascular tissue, pendrin gene ablation increased thoracic aorta contractile force in response to either α adrenergic agonists (phentolamine) or angiotensin II [96]. This greater contractile force was associated with increased myosin light chain kinase 20 abundance and was eliminated when an angiotensin type 1a receptor inhibitor, i.e. candesartan, was given in vivo [96]. This increased vascular contractility may also limit the hypotension that follows pendrin gene ablation. However, because pendrin expression in vascular tissue is very low, the changes in vascular tone we observed with pendrin gene ablation likely occurred as an indirect effect of pendrin gene ablation, such as through changes in circulating levels of angiotensin II and/or catecholamines [96].
Because previous studies have used global, embryonic pendrin (Slc26a4) gene ablation to study pendrin physiology, some of the phenotype observed in these mutant mice may have occurred due to developmental changes or from an extra-renal effect of pendrin gene ablation. Eliminating these potentially confounding variables will require the development of an inducible, tissue-specific pendrin null mouse.
Conclusions
Angiotensin II and aldosterone increase renal NaCl absorption in part by increasing apical plasma membrane pendrin abundance. While angiotensin II and aldosterone directly stimulate ENaC in principal cells, they also stimulate ENaC activity indirectly, by stimulating pendrin, which then increases ENaC abundance and function. In the kidney, pendrin regulates ENaC abundance and function, at least partly, by changing luminal HCO3− and ATP. However, pendrin is also expressed in tissues outside the kidney that also modulate blood pressure, such as the adrenal medulla. Whether pendrin changes blood pressure, at least in part, through its action outside kidney remains to be determined.
Acknowledgments
This work was supported by NIH grant DK 104125 (to S. M. Wall). I thank Dr. Peter Kopp for his suggestions.
Notes
Conflicts of interest
The author has no conflicts of interest to declare.