Epigenetic modifications and diabetic nephropathy
Article information
Abstract
Diabetic nephropathy (DN) is a major complication associated with both type 1 and type 2 diabetes, and a leading cause of end-stage renal disease. Conventional therapeutic strategies are not fully efficacious in the treatment of DN, suggesting an incomplete understanding of the gene regulation mechanisms involved in its pathogenesis. Furthermore, evidence from clinical trials has demonstrated a “metabolic memory” of prior exposure to hyperglycemia that continues to persist despite subsequent glycemic control. This remains a major challenge in the treatment of DN and other vascular complications. Epigenetic mechanisms such as DNA methylation, nucleosomal histone modifications, and noncoding RNAs control gene expression through regulation of chromatin structure and function and post-transcriptional mechanisms without altering the underlying DNA sequence. Emerging evidence indicates that multiple factors involved in the etiology of diabetes can alter epigenetic mechanisms and regulate the susceptibility to diabetes complications. Recent studies have demonstrated the involvement of histone lysine methylation in the regulation of key fibrotic and inflammatory genes related to diabetes complications including DN. Interestingly, histone lysine methylation persisted in vascular cells even after withdrawal from the diabetic milieu, demonstrating a potential role of epigenetic modifications in metabolic memory. Rapid advances in high-throughput technologies in the fields of genomics and epigenomics can lead to the identification of genome-wide alterations in key epigenetic modifications in vascular and renal cells in diabetes. Altogether, these findings can lead to the identification of potential predictive biomarkers and development of novel epigenetic therapies for diabetes and its associated complications.
Diabetes and metabolic disorders have reached epidemic proportions worldwide and are major risk factors for several long-term complications, including progressive renal failure and chronic kidney disease, termed diabetic nephropathy (DN). More than 40% of patients with diabetes progress to DN, a major cause of end-stage renal disease (ESRD). In addition, patients with DN are more susceptible to macrovascular diseases such as atherosclerosis, hypertension, and stroke, leading to a higher mortality rate among diabetic patients with ESRD than among those without ESRD. DN is pathologically characterized by renal glomerular hypertrophy, expansion of mesangial and tubular compartments, accumulation of extracellular matrix in multiple renal cells, inflammatory cell infiltration, and podocytopenia associated with foot process effacement. These factors lead to glomerulosclerosis, tubular atrophy, and podocyte apoptosis/dysfunction. Clinically, DN is identified by albuminuria, rising creatinine levels, and aberrant glomerular filtration rates. Signal transduction mediated by key factors associated with diabetes, such as high glucose (HG), advanced glycation end products (AGEs), oxidized lipids, proinflammatory cytokines, and local growth factors including transforming growth factor-β1 (TGF-β) and angiotensin II (Ang II), can lead to renal hypertrophy and fibrosis through various biochemical mechanisms [1], [2], [3], [4], [5], [6].
Several treatment strategies are available to cure DN or reduce its progression. These include modalities used to suppress the renin–angiotensin–aldosterone system and control blood glucose levels. However, diabetic patients are still reaching ESRD at an alarming rate. Moreover, clinical and experimental studies have shown that the risk and severity of diabetic complications, including DN, seem to persist even after glucose normalization, suggesting a “metabolic memory” of the prior exposure to HG [7], [8]. These findings imply that the current knowledge based on biochemical mechanisms, genetics, and proteomics research [6], [9], [10], [11], [12] does not explain the pathophysiology of DN completely. More than a decade after the human genome has been sequenced, it is becoming increasingly clear that changes in the DNA sequence and genetic mutations alone cannot fully explain the diversity of cellular responses and disease incidence in the general population. On the other hand, epigenetic mechanisms in chromatin can also regulate gene expression, cellular identity, phenotypic variations, and disease states without any alterations in the underlying DNA sequence. Epigenetics can be influenced profoundly by diet, lifestyle, and the environment—key factors associated with the etiology of diabetes [13], [14]. Thus, complex interactions between genes and the environment can play significant roles in common human diseases such as diabetes and related pathologies. Given the high incidence and debilitating nature of DN, it is imperative to evaluate additional mediatory factors and therapeutic targets. The current review focuses on epigenetics and the emerging evidence for epigenetic mechanisms in diabetes and its complications, with special emphasis on DN.
Epigenetics
The term “epigenetics” originally referred to the programmed changes during development that cannot be explained by conventional genetics [15]. Epigenetics typically refers to changes in gene expression and phenotype, including those conferred mitotically or meiotically, that occur without any alterations in the underlying DNA sequence. More recently, epigenetics is defined as “the structural adaptation of chromosomal regions so as to register, signal, or perpetuate altered activity states” to account for alterations in the chromatin state [15], [16]. Epigenetic signals regulate gene expression through chromatin remodeling, which in turn regulates chromatin structure and access to transcription factors and coregulators. Epigenetic control of gene regulation plays an important role in embryogenesis, cellular differentiation and development, cell identity, genomic imprinting, immune cell function, stem cell plasticity, transcriptional memory, and cellular responses to environmental signals [17], [18], [19]. Poor dietary choices and exposure to environmental pollutants can regulate epigenetic mechanisms to induce abnormal metabolic phenotypes that can be further compounded by genetic susceptibility [14], [20]. Furthermore, environmentally induced epigenetic modifications could lead to phenotypic changes that persist even long after the original exposure was removed. Several studies also demonstrated the transgenerational inheritance of epigenetic changes, thus highlighting the potential effect of maternal malnutrition or overnutrition on the fetus that can result in deleterious effects later in adult life, including predisposition to diabetes and associated complications [13], [21], [22]. However, whether low birth weight increases the risk for DN is still not clear [23]. Furthermore, while the role of genetic factors associated with DN has been examined extensively, the role of epigenetics is only recently being considered.
Epigenetic modifications in chromatinand the epigenome
Chromatin, the site of epigenetic modifications in the nucleus, is composed of chromosomal DNA tightly packaged into repeating units called nucleosomes. Each nucleosome is made up of 146 bp DNA, which is wrapped around an octamer protein complex consisting of dimers of core histone proteins H2A, H2B, H3, and H4 held together by a linker histone H1 [24]. Chromatin structure is dynamically regulated by epigenetic modifications between transcriptionally silent “heterochromatin” and active “euchromatin” states (Fig. 1). These include covalent post-translational modifications (PTMs) of nucleosomal histones as well as DNA methylomes (DNAMes), while noncoding RNAs such as microRNAs can also regulate gene expression by post-transcriptional epigenetic mechanisms. Epigenetic modifications provide docking sites for proteins/enzymes that alter chromatin structure, remodel nucleosomes, and erase or recruit other transcription regulators. Combinatorial effects of epigenetic modifications on promoters, enhancers, and other regulatory elements of the genome dictate gene expression patterns and the resultant phenotypes under normal and pathophysiological conditions [20], [21], [25]. Genome-wide patterns of these modifications collectively form the “epigenome,” which is cell-type specific, can be modulated by environmental cues, and can lead to aberrant gene expression associated with human diseases [13], [20], [26].
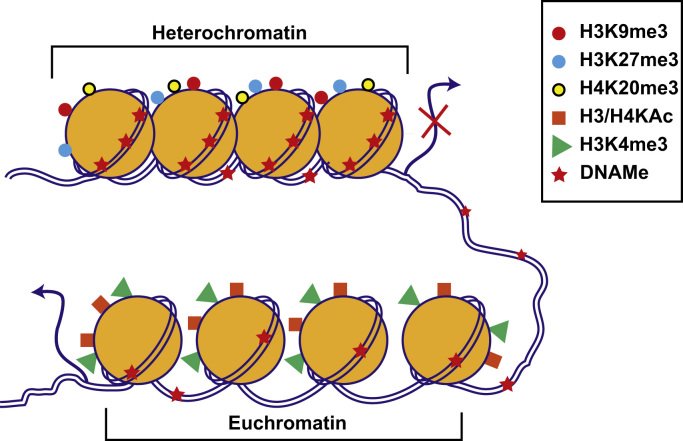
Chromatin structure and function. Chromatin is made up of repeating units of nucleosomes consisting of 146 bp DNA wrapped around dimers of four histone proteins (H2A, H2B, H3, and H4). The exposed amino-terminal tails of nucleosomal histones are subjected to post-translational modifications. Combinatorial effects of histone modifications and DNAMe regulate the chromatin structure between transcriptionally silent “heterochromatin” and active “euchromatin.” Enrichment of promoter DNAMe and histone modifications such as H3K9me3, H3K27me3, and H4K20me3 promote nucleosome condensation to repress transcription (heterochromatin). On the other hand, histone modifications H3 or H4 KAc (H3/H4KAc) and H3K4me promote open chromatin formation and increase accessibility to the transcription machinery, leading to active transcription (euchromatin). Other histone modifications such as Ser/Thr phosphorylation, ubiquitination and SUMOylation, and non-coding RNAs including microRNAs also regulate chromatin structure and function (not shown). Genome-wide patterns of DNAMe and histone modifications are referred to as the “epigenome.” Its response to internal and external signals regulates gene expression involved in diverse biological processes and disease conditions. DNAMe, DNA methylomes; KAc, lysine acetylation.
DNAMe, the most highly studied epigenetic modification, generally takes place on cytosines of CpG dinucleotides in regions known as “CpG” islands located in chromosomal DNA. DNAMe is catalyzed by DNA methyltransferases (DNMTs) that transfer methyl group from S-adenosyl-methionine to the 5′ position of cytosine (5′-methyl cytosine). In mammalian cells, DNMT3A and DNMT3B mediate de novo methylation while DNMT1 acts as a maintenance methyl transferase [18]. Promoter DNAMe generally leads to gene silencing via multiple mechanisms including inhibition of transcription factor binding, increasing nucleosome occupancy at transcriptional start sites, regulation of histone PTMs and chromatin remodeling, cross-talk with histone methyltransferases, acetylases and deacetylases, and recruitment of methyl binding proteins such as MeCP2 [18], [27]. Similar to other epigenetic modifications, DNAMe is now also known to be reversible [28], [29]. Non-CpG methylation has also been found in pluripotent cells such as embryonic stem cells or early embryos [30], [31], but it is not common in adult somatic tissues [32]. Although the role of non-CpG island methylation in mammals is largely unknown, recent studies have shown that it may also promote gene silencing and contribute to tumorogenesis [33]. DNAMe regulates important biological processes such as imprinting, differentiation, and X chromosome inactivation. It has been studied extensively because gene silencing of tumor suppressors and cell-cycle regulators via promoter hypermethylation has been associated with several cancers. Key epigenetic therapies based on DNAMe inhibitors have been developed for cancer [34], [35]. However, relatively less is known about the role of DNAMe in diabetes and its complications.
PTMs at chromatin histones also confer epigenetic changes in a dynamic fashion. Histone PTMs include lysine acetylation (KAc), lysine methylation (KMe), arginine methylation, serine/threonine phosphorylation, SUMOylation, Adenosine diphosphate (ADP)-ribosylation, and ubiquitination (Table 1) [17], [25]. In general, the amino acid residues in the exposed amino-terminal tails of nucleosomal histones are subjected to these PTMs; some PTMs within globular domain of histones have also been identified. Histone PTMs act in concert with each other, and this “histone code” can dictate gene transcription [17], [25]. Histone lysine acetylation (HKAc) and methylation (HKme) are the best studied and will be discussed in this review. HKAc is mediated by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs). On the other hand, histone lysine methyltransferases (HMTs) mediate KMe and are erased by histone lysine demethylases (HDMs). Recently, these enzymes have been renamed as lysine acetyltransferases, lysine methyltransferases, and lysine demethylases, because they can also modify lysines in nonhistone proteins [37].

Histone modifications such as H3K9/14 or H4K12/16 acetylation at gene promoters and enhancers are generally epigenetic marks for active gene transcription. HKAc is mediated by HATs such as CBP/p300 and SRC-1, which also act as coactivators. Acetylation of lysine residues in histone tails neutralizes the positive charge and destabilizes interactions between histones and DNA as well as nucleosome–nucleosome interactions. In addition, the acetylated residues act as docking sites for chromatin remodeling proteins, leading to chromatin relaxation and increased accessibility to the transcription machinery. HATs are quite nonselective and acetylate several lysine residues in both histone and nonhistone proteins [17], [38]. However, profiling approaches such as chromatin immunoprecipitation followed by next-generation DNA sequencing (ChIP-Seq) revealed differences in the genome-wide distribution of individual HATs [39]. Genome-wide approaches also showed that enhancers are usually associated with CBP and p300 along with histone lysine monomethylation (H3K4me1) [40], [41], [42].
HDACs remove HKAc marks and mediate gene repression. They are subdivided into four classes, I–IV, depending on their structure, function, and mode of action. HDACs restore the positive charge of histone lysines and promote chromatin condensation, resulting in decreased chromatin access and reduced rates of transcription. HDACs have multiple functions, which can be attributed to their low substrate specificity. Their actions can be regulated at the level of both gene expression and promoter-specific recruitment [43]. Studies in knockout mice revealed specific functions of HDACs in vivo, including embryonic stem cell differentiation and cardiovascular effects [44], [45], [46]. The function of HDAC in cancer cells has been studied extensively, and several HDAC regulators are being used to treat cancers [35], [47].
HKme is very interesting since it can be associated with either active or repressed genes depending on the modified lysine residue (Table 1). Furthermore, lysine residues can be mono (me1), di (me2), or tri (me3) methylated, adding another level of complexity. H3K4me, H3K36me, and H3K79me are usually associated with active promoters and gene bodies, while H3K9me, H3K27me, and H4K20me promote transcription repression. In contrast, demethylation of H3K4me can inhibit transcription, whereas demethylation of H3K9me, H3K27me, and H4K20me marks can increase transcription. HMTs generally contain a SET domain that catalyzes methyltransferase activity, whereas demethylation activity in HDMs is mediated by amine oxidase-like domains or JmjC domains [25], [48]. HMTs and HDMs are highly specific to the lysine residue being modified and also to the extent of methylation, e.g., SUV39H1 can mediate H3K9me3 but not H3K9me1, Ezh2 mediates H3K27me3, and SET7/9 mediates H3K4me1/me2. On the other hand, lysine-specific demethylase 1 (LSD1), the first HDM to be identified, can erase H3K4me1/me2, while the JARID1 family of demethylases removes H3K4me3. Jmjd2a can erase both H3K9me3 and H3K36me3, while Jmjd3 is specific to H3K27me3 [17], [49] (Table 1). Recent high-throughput profiling of various histone PTMs associated with promoters, enhancers, and insulators led to the identification of such key regulatory elements genome wide [25], [42], [50], [51]. Transcription start sites and promoters are enriched with H3K4me3 and H3K9/14Ac, enhancers with H3K4me1, and gene bodies of actively transcribed genes with H3K36me3. Histone modifications at enhancers also confer tissue- and cell-specific transcription in conjunction with lineage- or cell-specific transcription factors [25], [42], [52], [53]. Histone modifications can also cross-talk with each other to regulate gene expression. Thus, H3K27me3 and H3K4me3 can antagonize each other, and promoters with these bivalent modifications play important roles in stem cell differentiation [54]. H3K9Ac and Ser10 phosphorylation generally inhibit H3K9me to promote gene expression [17]. Cross-talk between histone PTMs and DNAMe has also been documented. DNAMe acts in concert with repressive histone modifications such as H3K9me3 to recruit transcription repressors and silence gene expression [20], [55].
Regulation of histone-modifying enzymes plays a critical role in gene expression involved in key biological processes including differentiation, cancer, and diabetes and its complications [7], [8], [20], [34], [36], [56], [57]. In general, HKme is relatively more stable compared with other histone modifications. Therefore, similar to DNAMe, H3Kme might be involved in the inheritance of epigenetic signatures to confer transcriptional memory that lasts long even after the removal of physiological or pathological signals. Polycomb repressive complex (PRC) proteins, which mediate the repressive H3K27me3 mark, remained tightly bound to chromatin during DNA replication, indicating a potential epigenetic role for such histone marks in the maintenance of transcription memory or epigenetic transmission [58], [59]. Thus, alterations in HKme may contribute to the metabolic memory of transcription and persistent expression of pathologic genes implicated in diabetic complications [8], [57]. As discussed earlier, epigenetic marks altered by the environment can be inherited during both somatic and germ cell replications [60]. Evidence shows that DNAMe plays an important role in transgenerationally inherited obesity [61]. In addition, high-fat diets have been found to promote transgenerational transmission of adult-onset metabolic diseases in mice, suggesting an underlying epigenetic mechanism [62]. In epigenetic transmission, the role of DNAMe is more apparent than that of histone modifications, although evidence also exists for the latter via PRCs and histone recycling [59], [63]. In general, compared to genetic transmission, epigenetic transmission is more complex and needs further investigation, especially in the field of diabetes and its complications.
Metabolic memory and diabetes complications
Hyperglycemia has been implicated in the development of numerous diabetic complications including nephropathy. Patients with both type 1 diabetes (T1D) and type 2 diabetes (T2D) continue to be at higher risk for developing complications even several years after achieving normoglycemia. This phenomenon of “metabolic memory” was clearly demonstrated in clinical trials such as the Diabetes Control and Complications Trial (DCCT) and the follow-up Epidemiology of Diabetic Interventions and Complications (EDIC) study [7], [8], [64]. The DCCT showed that intensive insulin therapy to attain strict glycemic control in T1D patients led to much better control of various vascular complications than conventional therapy [65]. Subsequently, both standard and intensive treatment DCCT groups were placed on intensive insulin therapy and followed up long term in EDIC study. Interestingly, EDIC study showed that those previously assigned to standard/conventional therapy in DCCT continued to develop complications, such as nephropathy and retinopathy, at significantly higher rates than the previous intensive therapy group in DCCT [66]. Recent reports demonstrate continued protection and lower rates of cardiac, renal, and retinal complications; lower blood pressure; more favorable lipid profiles; and reduced long-term risk for impaired glomerular filtration rate (GFR) in the former DCCT intensive therapy group [67], [68], [69], [70]. Other clinical trials in T2D patients also demonstrated that a similar memory phenomenon, termed “legacy effect,” in those with lower fasting blood glucose and strict glycemic control resulted in lower rates of vascular complications [71], [72], [73]. These studies clearly demonstrated the benefits of strict glycemic control and the fact that a memory of prior exposure to hyperglycemia might play a significant role in the subsequent risk of diabetic complications including nephropathy. However, mechanisms of metabolic memory are not clear and remain a major obstacle in the prevention or treatment of diabetic complications. As discussed later, recent studies have evaluated a role for epigenetic mechanisms in metabolic memory.
Several experimental cell culture and animal models have been developed to understand the mechanisms involved in metabolic memory. Endothelial cells (ECs) cultured in HG for short time periods continued to exhibit enhanced oxidative stress, and profibrotic and inflammatory genes even after return to normal glucose [74], [75], [76]. Vascular smooth muscle cells (VSMC) derived from the aortas of T2D db/db mice exhibited enhanced proinflammatory responses even after culturing ex vivo for a few weeks [77], [78]. In T1D dogs, islet transplantation performed 12 weeks after the development of diabetes did not prevent progression to retinopathy, compared to that after 6 weeks [79]. Similarly, in T1D rats, reinstitution of glycemic control following several weeks of poor glycemic control did not reverse inflammation or progression to retinopathy and nephropathy [80], [81], [82], [83]. Thus, both clinical and experimental evidences suggest that prior exposure to hyperglycemia can have long-lasting deleterious effects even after subsequent glycemic control. Other related factors associated with hyperglycemia and diabetes such as AGEs may also contribute to metabolic memory, because AGEs can modify various cellular proteins and enzymes to cause long-lasting changes in their functions, or can activate multiple signaling pathways via the receptor for AGE (RAGE) to induce inflammatory genes [84], [85]. Furthermore, dyslipidemia and insulin resistance associated with T2D may also be involved. It is likely that these factors acting independently or cumulatively can lead to changes in epigenetic histone PTMs and DNAMe in target cells, thus contributing to metabolic memory (Fig. 2).
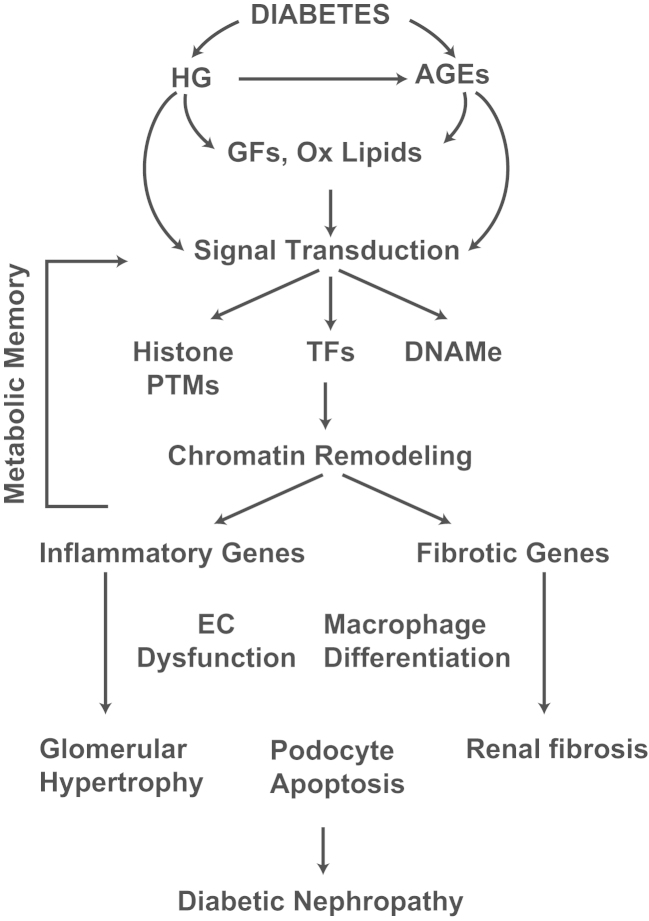
Diabetic nephropathy and metabolic memory. Signal transduction mediated by factors associated with the diabetic milieu (HG, AGEs, growth factors, and oxidized lipids) induces alterations in epigenetic modifications (histone PTMs and DNAMe) and changes in chromatin remodeling. This leads to pathologic gene expression (inflammatory genes and fibrotic genes) in collaboration with key TFs in monocytes/macrophages, vascular and renal cells. Persistence of these epigenetic modifications even after normalizing glucose levels can play an important role in “metabolic memory” and gene expression implicated in long-lasting diabetic complications including nephropathy. AGEs, advanced glycation end products; DNAMe, DNA methylomes; EC, endothelial cell; GF, growth factor; HG, high glucose; Ox lipids, oxidized lipids; PTM, post-translational modification; TF, transcription factor.
DNAMe and diabetes complications
DNAMe is the most stable and heritable epigenetic modification and is known to play an important role in normal as well as pathophysiological processes. However, its involvement in diabetes and its complications is less well studied. Early studies with mouse models showed that regulation of the Agouti gene by DNAMe plays a key role in the development of coat color, diabetes, and obesity. Interestingly, these studies also demonstrated the influence of external factors such as maternal diet on epigenetic processes and the transgenerational inheritance of DNAMe [60], [86]. In a model of intrauterine growth retardation (IUGR) in rats, it was found that the development of diabetes was associated with promoter hyper-DNAMe and inhibition of the islet-specific transcription factor Pdx1 that regulates insulin gene expression in pancreatic β cells. DNAMe was also found to be related to downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) and Igf1r in pancreatic islets and skeletal muscles of T2D animals and patients [87], [88], [89]. PGC-1α downregulation led to the inhibition of insulin expression and mitochondrial biogenesis [87], [88]. Furthermore, in another study, insulin promoter DNAMe in islets of T2D patients was correlated with lower insulin levels and elevated HbA1c levels [90]. Another recent study on monozygotic twins reported a 10% increase in global DNAMe at Alu repetitive elements in peripheral blood leukocytes, which showed significant correlation with insulin resistance [91]. Genetic association studies have identified single nucleotide polymorphisms in the fat mass and obesity-associated (FTO) gene region with a risk of adiposity. Functional studies showed that FTO controls feeding behavior and energy expenditure [92]. Interestingly, recent studies in T2D patients showed a small but significant decrease in DNAMe at a CpG site in the first intron of the FTO gene [93]. Furthermore, this hypomethylation was also observed in healthy individuals who later progressed to impaired glucose metabolism. These unique findings suggest a correlation between epigenetic DNAMe variations and predisposition to T2D [93].
Studies, reported to date, investigating kidneys of diabetic animal models or renal cells treated in vitro with HG have not demonstrated significant changes in DNAMe [94], [95], [96]. On the other hand, whole blood genomic DNA from T1D patients with DN exhibited differential DNAMe patterns at 19 genes including UNC13B, relative to those without nephropathy [97]. Interestingly, UNC13B was correlated with DN in genetic studies, suggesting an association between DNAMe and DN susceptibility. Several differentially methylated genes were also identified in DNA extracted from saliva of ESRD patients compared to patients with chronic kidney disease who did not progress to ESRD [98]. However, relevance of these findings to DN awaits further investigation. It is likely that high-throughput approaches using genomic DNA and target tissue biopsies from various clinical cohorts or animal models will clarify the relationship between DNAMe and diabetic complications including nephropathy. In this connection, it is noteworthy that a recent genome-wide profiling study using next-generation sequencing demonstrated significant changes in DNAMe in HG-treated ECs at several genes related to diabetes and its complications. Reciprocal relations with histone acetylation were also demonstrated [99].
Histone modifications, diabetes complications, and metabolic memory
Progress in the study of histone PTMs has grown by leaps and bounds in the field of cancer. However, it is still in its infancy in the field of diabetes. Recent studies have shed some insights into the role of histone modifications in diabetes, diabetic complications, and metabolic memory. Histone PTMs have been implicated in the regulation of insulin expression by Pdx1 in pancreas. In response to elevated glucose levels, Pdx1 could recruit the HAT coactivator p300 and HMT SET7 to the insulin promoter, which could promote chromatin relaxation by increasing H3K9/14Ac and H3K4me1/2, respectively, to enhance insulin transcription. In contrast, Pdx1 could recruit HDACs to inhibit insulin expression when glucose levels were low [100], [101], [102]. Interestingly, it was found that IUGR reduces insulin gene expression by inhibiting Pdx1 expression via alterations in promoter H3KAc, H3K4me3, and DNAMe in the offspring [103]. H3K9me2 and H3K4me2, as well as the related enzymes SETDB1 (H3K9 methyl transferase) and LSD1 (H3K4 demethylase), were reported to play critical roles in adipocyte differentiation [104]. More direct evidence for histone modifications in diabetes was suggested in a report, which showed that mice deficient in Jhdm2a, an H3K9me2 demethylase, exhibited features of the metabolic syndrome [105]. A recent genome-wide analysis of histone modifications revealed a global snapshot of the islet chromatin, and several islet-specific enhancers were identified. Interestingly, many of them were located in genetic loci previously associated with T2D [106]. HDACs were also shown to regulate the acetylation of several transcription factors involved in adipocyte differentiation, suggesting an important role for non-HKAc in metabolic disorders [107], [108]. However, the role of HDACs in epigenetic mechanisms involved in diabetes complications and the potential use of HDAC inhibitors are not well studied.
Aberrant promoter histone modifications can be triggered by HG and other diabetogenic agents to affect adversely the expression of key target genes associated with complications. Several studies have examined the role of histone PTMs in the regulation of inflammatory gene expression in ECs, monocytes, VSMCs, and renal mesangial cells (MCs), all of which are key cell types involved in diabetes complications [6], [57], [109], [110], [111], [112]. Endothelial dysfunction, increased oxidant stress, and reduced nitric oxide are major features of diabetic micro- and macrovascular complications. In particular, this is associated with decreased levels or activity of the endothelial nitric oxide (eNOS) enzyme and is exemplified in studies involving eNOS knockout mice [109]. Diabetes reduces NO bioavailability, resulting in increased EC proliferation, inflammatory gene expression, VSMC activation, and macrophage infiltration, which are the key factors in the development of DN [109], [110]. EC-specific expression of eNOS involves formation of an open chromatin near the eNOS promoter marked by increased H3/H4KAc and H4Kme [113], [114]. Interestingly, hypoxia, a known mediator of EC dysfunction and a potential risk factor for DN [115], also inhibits eNOS expression through reversal of these promoter-specific histone modifications [116]. Other studies have shown that gene expression in monocytes and macrophages is also regulated by histone modifications, which play key roles in inflammation and macrophage polarization [57], [117]. JMJD3, an H3K27me3 demethylase, and HDAC11 appear to play important regulatory roles in these events [118], [119], [120]. HG-induced inflammatory gene expression was associated with increased promoter H3/H4KAc, and enhanced recruitment of HATs CBP/P300 as well as the proinflammatory transcription factor NF-κB in THP-1 monocytes. Furthermore, monocytes from diabetic patients exhibited enhanced H3KAc at the promoters of key inflammatory genes, confirming in vivo relevance [121]. SET7, an H3K4 methyltransferase, was implicated in NF-κB-mediated inflammatory gene expression in TNF-α-treated THP-1 monocytes, and in macrophages from diabetic mice [122]. Profiling studies using ChIP linked to microarrays (ChIP-on-chip) revealed key differences in the patterns of repressive (H3K9me2) and active (H3K4me2) histone modifications at coding regions of HG-regulated genes in THP-1 monocytes, with in vivo relevance being shown in primary monocytes from diabetes patients [123]. Furthermore, comparative ChIP-on-chip epigenome profiling of lymphocytes from T1D patients with those from normal control volunteers revealed significant alterations in H3K9me2 at a subset of genes related to T1D and closely associated with autoimmune functions and inflammation [124], further demonstrating the role of histone modifications in diabetes and its complications. Recent ChIP-on-chip studies with chromosome 6 tiling arrays in monocytes of T1D patients versus normal control individuals revealed key variations in H3K9Ac at the upstream enhancer regions of two HLA genes whose single nucleotide polymorphisms (SNPs) are known to be closely related with T1D [125], suggesting potential connections between genetic SNPs and surrounding epigenetic marks in the etiology of diabetes.
Emerging evidence supports a role for histone modifications in the regulation of key genes associated with DN. In a rat model of T2D-induced DN, hyperglycemia in the kidneys was associated with reduced levels of active chromatin marks H3KAc and Ser10 phosphorylation at the fibrillin 1 and collagen type III alpha1 gene promoters [126]. Changes in global histone modifications were seen in the kidneys of uninephrectomized diabetic db/db mice [127]. Furthermore, HDAC2 activity was increased in kidneys of diabetic animals, and inhibition of HDAC activity blocked diabetes-induced fibrotic gene expression [128]. Recently, evidence of epigenetic mechanisms in HG- and TGF-β1-induced fibrotic gene expressions in renal MCs was demonstrated for the first time [129]. In these studies, the induction of key profibrotic genes by HG and its downstream effectors TGF-β1 was associated with increases in activation marks (H3K4me-1, H3K4me-2, and H3K4me-3) and decreases in repressive marks (H3K9me-1 and H3K9me-2) at these gene promoters. Furthermore, HG and TGF-β1 could induce the expression of SET7, while SET7 gene silencing inhibited TGF-β1-induced expression of fibrotic genes. Interestingly, pretreatment with TGF-β1 antibodies significantly inhibited HG-induced profibrotic gene expression and alterations in histone modifications at their promoters [129]. These results demonstrate the involvement of histone PTMs in HG- and TGF-β1-induced genes relevant to DN (Fig. 3).

Role of TGF-β in HG-mediated histone modifications and fibrotic gene expression in mesangial cells. HG and its downstream effector TGF-β promote open chromatin formation at fibrotic gene promoters through inhibition of repressive marks (H3K9me) and increasing activation marks (H4Kme) via regulation of HMTs such as SET7. This leads to an increase in chromatin access to transcription TFs and in the expression of fibrotic genes such as PAI-1, Col1a1, and CTGF. These HG-induced alterations in epigenetic mechanisms and profibrotic gene expression can be blocked by TGF-β antibodies. Potential role of other histone modifications such as H3K9Ac and coactivator HATs is also shown. HAT, histone acetyltransferase; HG, high glucose; HMT, histone methyl transferase; TF, transcription factor; TGF-β, transforming growth factor-β1.
Role of histone modifications was also demonstrated in some nondiabetic kidney diseases. Thus, reports showed that aldosterone can reverse Dot1a-mediated promoter repressive H3K79me mark to increase the expression of the epithelial Na (+) channel alpha (ENaCα) gene in the renal collecting ducts [130], [131]. In other studies, increased expression of inflammatory genes such as MCP-1 in a model of renal ischemic injury was associated with increased H3K4me3 and recruitment of the chromatin remodeling enzyme Brahma-related gene-1 (BRG1) at these gene promoters [132]. Changes in repressive histone PTMs were associated with collagen III gene expression in aging-induced nephropathy [133]. Notably, epigenetic mechanisms have been implicated in renal development and maintenance of podocyte-differentiated function [134], [135]. Together, these data demonstrate that histone PTMs are involved in the regulation of genes associated with chronic as well as acute renal injuries, and even with renal development. However, such candidate-gene-based approaches can only provide a glimpse of a complex epigenetic landscape. Genome-wide high-throughput profiling approaches can help provide valuable information about the state of the epigenome in DN.
Recent studies using cell and animal models have suggested that histone modifications might be associated with metabolic memory. In one study, there was enhanced inflammatory gene expression in VSMCs cultured from T2D db/db mice for several passages relative to those from genetic control db/+ mice, and this was associated with persistently reduced H3K9me3 and occupancy of corresponding methyl transferase Suv39h1 at inflammatory gene promoters [78]. Furthermore, there was also reduced levels of Suv39h1 in the diabetic db/db VSMCs, which was found to be due to an increased expression of the microRNAs miR-125b that targets Suv39h1 [136]. The miR-125b mimics could reduce Suv39h1 levels, upregulate inflammatory genes in nondiabetic cells, and also promote monocyte binding in db/+ VSMCs to induce a diabetic phenotype. This demonstrates a novel cross-talk between microRNAs and epigenetic mechanisms in diabetic vascular inflammation and metabolic memory. In another model of metabolic memory, short-term exposure of ECs to HG led to persistently increased expression of p65 (active subunit of NF-κB) and inflammatory genes for several days even after returning to normal glucose. This was associated with higher promoter levels of H3K4me1 and occupancy of the corresponding SET7 H3K4 methyl transferase [76], [137]. A key role for oxidant stress in promoting metabolic memory in ECs was suggested in this and other studies [74], [76], [99]. Interestingly, in a rat model of diabetic retinopathy and metabolic memory, retinal ECs exhibited reduced expression of sod gene that codes for the protective antioxidant protein superoxide dismutase. This was associated with increased levels of the repressive mark H4K20me3 and occupancy of the corresponding methyltransferase Suv420h at the sod gene promoter, leading to repression of superoxide dismutase [83]. Together, these studies support a key role for histone modifications in metabolic memory of diabetic vascular complications that seem to persist despite glycemic control (Fig. 2). Since renal MCs, tubular epithelial cells, ECs, and podocytes are also affected by HG and diabetes, similar mechanisms in these cells in vivo might also contribute to metabolic memory of DN. Moreover, it has recently been speculated that epigenetic mechanisms may also regulate hypertension, which is associated with both diabetic and nondiabetic renal diseases and can persist across generations [138].
Resources and technologies for epigenomics research in diabetes and its complications
The National Institutes of Health Roadmap Epigenomics Program is focused on mapping the locations and characterizing the functions of epigenetic modifications in normal and diseased states using numerous cell types and tissues. These epigenomic maps are being made available to the entire scientific community (http://www.ncbi.nlm.nih.gov/epigenomics) and will be a tremendous resource for future research. Recent advances in next-generation sequencing and related technologies as well as in bioinformatics tools have accelerated epigenomics research significantly [25], [139], [140]. These include ChIP-on-chip and ChIP-Seq to study genome-wide patterns of histone modifications [25], [57], [139], [141]. DNAMe is traditionally studied using bisulfite conversion of 5′-methyl cytosine or approaches to enrich methylated DNA [142], [143]. These are coupled to microarrays or next-generation DNA-Seq analysis to determine genome-wide distribution of DNAMe [57], [144]. Novel features of the epigenome, including key regulatory elements/regions, have been uncovered from the combinatorial and integrative analyses of genome-wide patterns of histone PTMs and DNAMe along with transcriptome analyses of gene expression patterns [25], [42], [50], [51], [140], [145]. These technologies are gradually being utilized to decipher the epigenomes of key target cells involved in diabetes and its complications. Utilization of these genome-wide profiling technologies to study renal biopsies or genomic DNA from patients with DN or other renal diseases relative to normal control individuals can yield significant information regarding the epigenome of normal versus diseased kidneys, which in turn could open the doors for the development of novel therapeutic opportunities. However, it is also important to consider conducting such studies on cells or tissues that manifest the disease, since the epigenome is cell-type specific. This could pose a challenge in renal research due to the multicellular nature of the kidney.
Summary
Increasing evidence supports the critical role of hyperglycemia and downstream mediators such as AGEs, Ang II, and TGF-β in signaling, molecular, and chromatin-based mechanisms in the development of metabolic memory and diabetic complications (Fig. 2). However, diabetes leads to multiorgan dysfunction and is associated with several physiological abnormalities, including insulin resistance and dyslipidemia (T2D) and autoimmune disorders (T1D). It is further complicated by dietary choices and the lifestyles. Although numerous studies have examined the role of genetic variants in the pathology of diabetes complications such as DN, very few candidate loci with functional associations have been uncovered. Given the strong influence of environmental factors, the additional role of epigenetics is worth considering. Furthermore, metabolic memory still remains a major obstacle in the treatment and prevention of diabetes complications. Recent advances in high-throughput genomic sequencing approaches should make it feasible to better understand the epigenome state in diabetes and related complications such as DN. Importantly, they might reveal key differences in the epigenome between patients who progress to DN and other complications and those who do not. Unlike genetic changes, epigenetic changes are reversible, which thus provides a window of opportunity for therapeutic development. Several epigenetic regulators such as inhibitors of DNAMe, HDACs, HATs, and DNMTs are already being used for cancer treatment, and some are being evaluated for vascular disorders such as restenosis [35], [47], [146], [147]. Recent studies showing reversal of epigenetic modifications associated with islets [103], ECs [113], monocytes [148], and renal cells [128], [129], [149] indicate that epigenetic therapy for diabetic complications is a distinct possibility. Further work in this rapidly expanding area of research could advance our knowledge of epigenetic mechanisms involved in the pathogenesis of DN and other diabetic complications, which in turn could lead to new therapeutic strategies.
Conflict of interest
None declared.
References
Acknowledgments
The authors gratefully acknowledge funding from the National Institutes of Health (NIDDK and NHLBI), the Juvenile Diabetes Research Foundation, and the American Diabetes Association.