



Cho et al. [1] conducted a study in Korea that is considered the first attempt to utilize plasma globotriaosylsphingosine (lyso-Gb3) concentration as a screening tool for Fabry disease (FD) in chronic kidney disease (CKD) patients, including those undergoing dialysis. The authors found a prevalence rate of FD in these patients to be 0.11%, consistent with rates reported in other studies. Previous research has indicated prevalence rates of approximately 0.2% to 0.95% for CKD patients and 0% to 1.69% for male hemodialysis patients [2,3]. Utilizing lyso-Gb3 for screening purposes proves to be a valuable method for identifying FD cases within the CKD population with unknown etiology.
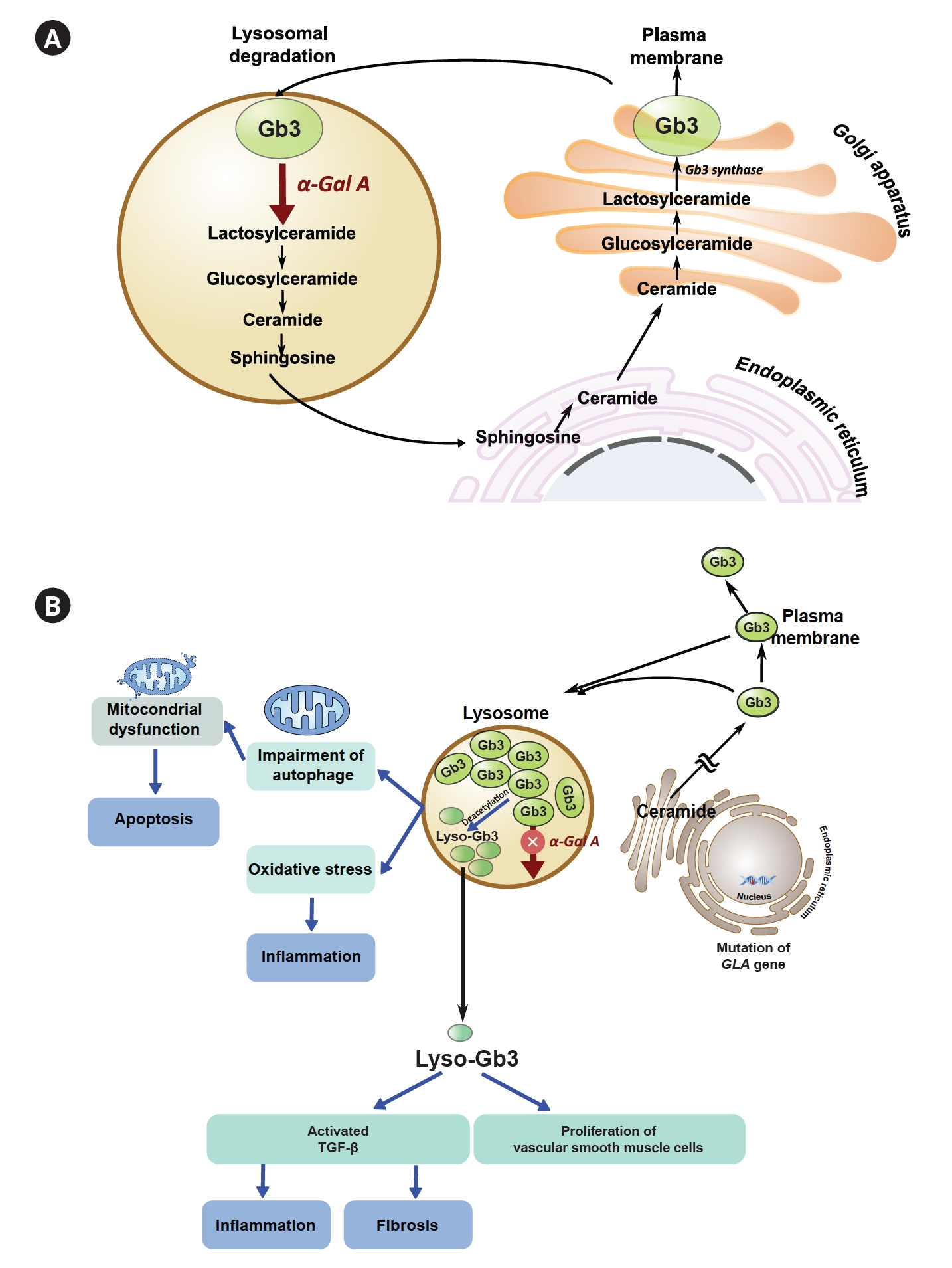
FD is a rare X-linked lysosomal disorder caused by a genetic mutation in the GLA gene. This gene codes for the enzyme α-galactosidase A (α-Gal A), and its deficiency leads to the accumulation of complex glycosphingolipids, especially globotriaosylceramide (Gb3) and its metabolites such as lyso-Gb3, in various tissues and organs. Fig. 1A shows the synthesis from ceramide to Gb3, which is useful in cellular membranes. The pathogenesis of Gb3 and lyso-Gb3 in FD patients depicted in Fig. 1B, Gb3 and lyso-Gb3 accumulation in various tissues leads to impairment of autophagy, which, in turn, induces mitochondrial dysfunction, increased apoptosis, and inflammatory immune responses [4]. Studies have demonstrated that Gb3 accumulation in various tissues leads to a dose-dependent increase in reactive oxygen species production in cultured vascular endothelial cells from individuals with FD [5]. Additionally, an in vitro study has shown the production of transforming growth factor beta 1 when normal podocytes are exposed to lyso-Gb3 [6] and plasma lyso-Gb3–induced proliferation of vascular smooth muscle cells, which leads to inflammation and fibrosis [7]. Furthermore, exposure to lyso-Gb3 has been shown to cause podocyte injury, glomerular fibrosis, and inflammation [6].
The clinical features of FD are diverse and can vary in severity depending on the age of onset and the degree of enzyme deficiency. Typical clinical features of this disorder include angiokeratoma, corneal verticillata, hypohidrosis, peripheral neuropathy, recurrent diarrhea, abdominal pain, ventricular hypertrophy, progressive kidney dysfunction, and cryptogenic stroke. FD phenotypes can be classified as classic, characterized by typical multiorgan involvement, and later-onset (or variant), predominantly affecting specific organs. Classic phenotype males showed that α-Gal A activity may be very low (or absent), develop symptoms earlier, and show a more severe presentation. But the pattern of organ-specific X-chromosome inactivation in heterozygous females showed that α-Gal A activity may be low-normal or variably deficient with variable symptoms [8]. Late-onset FD patients may present with nonspecific symptoms and signs.
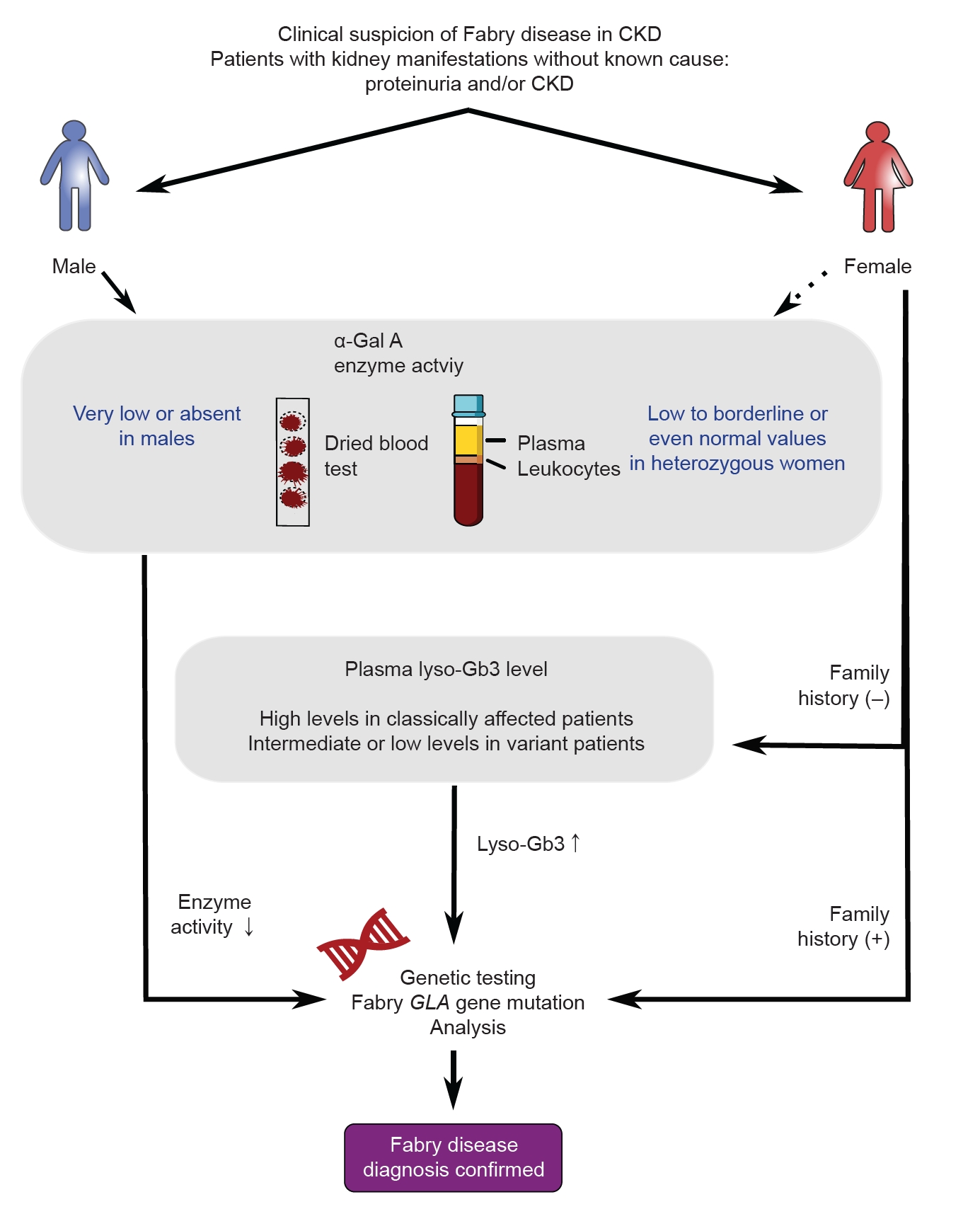
Since FD can potentially be a cause of CKD with an unknown origin, it is recommended to test any patient suspected of having FD (Fig. 2). However, there may be differences in how this condition presents itself in males and females. In males suspected of having FD, it is important to measure α-Gal A activity. An α-Gal A activity level of less than 1% highly suggests classic FD. The diagnosis of FD involves measuring α-Gal A activity in plasma, dried blood spots (DBS), and leukocytes. While plasma is commonly used, some cases may yield inconclusive results, highlighting the need for accurate sample processing. DBS offers similar performance with easier collection and a lower cost, although it may lack accuracy in certain cases. Leukocytes provide a definitive diagnosis but require complex processing and are more expensive. Therefore, careful sample processing and selection are crucial, considering the advantages and disadvantages of plasma, DBS, and leukocytes.
On the other hand, in females suspected to have FD, the α-Gal A activity can vary and may fall within the normal range. Genetic testing is a confirmatory and primary screening method for females; however, it can be costly. Therefore, there is a need for biomarkers that can be used for FD screening and diagnosis in individuals with normal or borderline α-Gal A activity. Lyso-Gb3, a hydrophilic deacylated form of Gb3, has emerged as a promising biomarker. It is detected at high levels in the plasma of FD patients and shows superiority over Gb3 as a diagnostic and prognostic biomarker [9]. Lyso-Gb3 levels increase early in disease progression, respond more rapidly than Gb3, and have been linked to clinical events and disease burden [7]. Because it is a smaller molecule, lyso-Gb3 may freely reach cells, which enables it to be detected more readily [9]. Lyso-Gb3 levels vary among individuals with different disease severity and genotypes, indicating their potential as an indicator of disease severity and different phenotypic presentations [9]. Even when typical symptoms of FD are present, diagnosing the condition can be challenging if the underlying genetic abnormality is unknown. Understanding the characteristics of lyso-Gb3, as mentioned earlier, would be beneficial for diagnosing such patients as well. Assessing plasma lyso-Gb3 levels can help evaluate disease severity and aid in the diagnosis of patients with genetic variants of unknown significance [10]. Hence, if there is a family history of FD in females, genetic testing should be performed next. However, in the absence of family history, it may be considered to perform genetic testing if there is an increase in lyso-Gb3 levels.
To summarize, early detection of FD is essential for improving patient outcomes and safeguarding the health of their family members. Therefore, it is crucial for physicians to have an effective screening tool. The lyso-Gb3 test, conducted by the researchers, is a laboratory test currently available for experimental purposes in domestic settings. The study’s findings underscore the significant role of lyso-Gb3 in FD screening and the establishment of screening criteria within the country, especially for females and late-onset FD. It is hoped that this research will pave the way for broader insurance coverage of lyso-Gb3 as a diagnostic technique for FD screening in the future.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









