Application of C5 inhibitors in glomerular diseases in 2021
Article information

Abstract
The complement pathway is an essential mechanism in innate immunity, but it is also involved in multiple pathologies. For kidney diseases, strong evidence of a dysregulation in the alternative pathway in atypical hemolytic uremic syndrome (aHUS) led to the use of eculizumab, the first anti-C5 inhibitor available in clinical practice. Intensive fundamental research resulted in the development of subsequent new drugs, such as long-acting C5 inhibitors, oral medications, or antagonists of C5aR, the receptor for C5a. New data in the domain of C5-inhibition in glomerular diseases are still limited and mainly focus on 1) the efficacy of ravulizumab, a long-acting C5 inhibitor in aHUS, and 2) the use of avacopan, a C5aR antagonist, in antineutrophil cytoplasmic antibody vasculitis. Several new studies ongoing or planned for the next few years will evaluate the efficacy of C5 inhibition in secondary thrombotic microangiopathy, C3 glomerulopathy, membranous nephropathy, or immunoglobulin A nephropathy.
Introduction
Anticomplement therapies were first developed to treat paroxysmal nocturnal hemoglobinuria (PNH), a clonal hematopoietic stem cell disorder, which results in the absence of CD59, a regulator of the complement system, on the surfaces of affected red blood cells. Patients with PNH experience a complement-dependent intravascular hemolysis that is mostly resolved with the administration of eculizumab, a humanized anti-C5 antibody [1]. This was the first major breakthrough indication for this drug.
The second breakthrough occurred when eculizumab was introduced in the field of nephrology. After it was shown that there is a dysregulation of the alternative pathway in atypical hemolytic uremic syndrome (aHUS), eculizumab was used to treat the disease. It showed impressive results, with a reduction in end-stage kidney disease from 50% at 1 year in historical cohorts to 6%–15% after treatment [2–5].
By blocking the terminal part of the complement system, the innate immunity is partially inactivated, especially against encapsulated bacteria. Although these therapies are then a risk factor for invasive meningococcal infections, infection can be efficiently prevented by vaccination and prophylactic antibiotherapy.
This review will focus on the most recent findings concerning the use of anti-C5 drugs in glomerular disease. Several other complement inhibitors (inhibitors of the lectin pathway, factor B, etc.) have been developed and are currently being evaluated—or will be assessed in the future—for glomerular diseases. However, these will not be discussed in the present review.
Complement system: basics
The complement system can be activated through three pathways: the classical pathway, the mannose-binding lectin pathway, and the alternative pathway (Fig. 1). Of these, the classical pathway is activated after recognition of immune complexes by C1q, and the lectin pathway is activated mainly by microbial surfaces, whereas the alternative pathway is spontaneously activated by the phenomenon of “tick-over.” The alternative pathway amplifies the response of the first two pathways or can be activated by properdin.
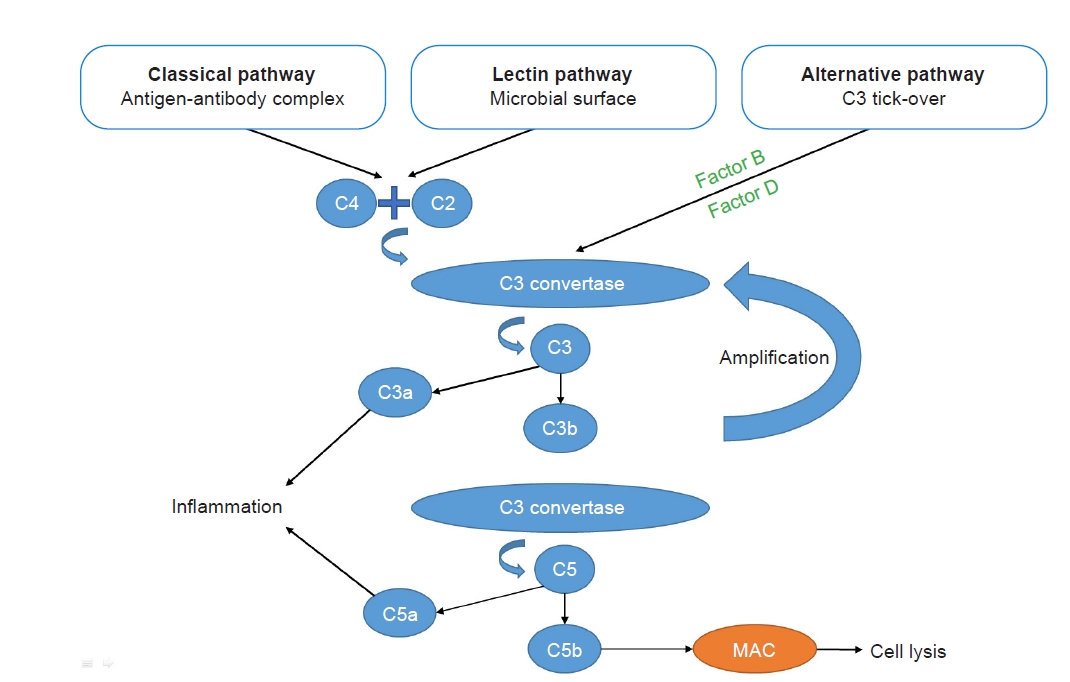
Complement system: a brief summary.
The complement system can be activated through three pathways: the classical pathway, the mannose-binding lectin pathway, and the alternative pathway. The classical pathway is activated following recognition of immune complexes, and the lectin pathway is activated mainly by microbial surfaces, whereas the alternative pathway is spontaneously activated by the phenomenon of “tick-over.” The alternative pathway amplifies the response of the first two pathways. These pathways lead to the formation of a C3 convertase, which cleaves C3 into C3a and C3b. C3b is then incorporated to form a C5 convertase, which cleaves C5 into C5a and C5b. C3a and C5a are proinflammatory molecules. C5b, together with C6, C7, C8, and C9, leads to the formation of the membrane attack complex (MAC), which results in cells lysis.
These pathways lead to the formation of a C3 convertase (C4bC2a for the classical and lectin pathways or C3bBb for the alternative pathway) that cleaves C3 into C3a and C3b. C3b is then incorporated to form a C5 convertase (C4bC2aC3b for the classical and lectin pathways or C3bBbC3b for the alternative pathway) that cleaves C5 into C5a and C5b. C3a and C5a, called anaphylatoxins, are proinflammatory molecules that, following ligation to their inflammatory cell receptors, trigger a release of proinflammatory cytokines and vasoactive agents. Meanwhile, C3b itself promotes opsonization. Together with C6, C7, C8, and C9, C5b leads to the formation of the membrane attack complex, resulting in cell lysis (endothelial cells, bacteria, etc.) [6].
The complement system, particularly the spontaneous tick-over, requires tight control, which is assumed by inhibitors such as factor H, factor I, monocyte chemotactic protein (MCP), and CD55. These proteins are involved at multiple checkpoints to contain the reaction [6].
Glomerular deposition of the membrane attack complex has been reported in a large proportion of patients with various kidney diseases but is located variably depending upon the disease, such as along the capillary wall in membranous nephropathy and lupus; in the mesangium in immunoglobulin A (IgA) nephropathy and lupus; or throughout the glomerulus in C3 glomerulopathy (C3G), aHUS, and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. It is tempting to test the efficacy of anti-C5 therapies in these conditions, but the causal role of the complement system remains unclear in a majority of these diseases (Table 1) [7].

Summary of existing or planned glomerular disease studies designed to test the efficacy of C5 inhibition
Most of the therapies developed thus far to target C5 inhibit the cleavage of C5 into C5a and C5b, but other drugs have a different mechanism of action—for example, avacopan blocks the C5a receptor and cemdisiran inhibits C5 production in hepatocytes. The properties of these C5 inhibitor drugs are shown in Fig. 2.
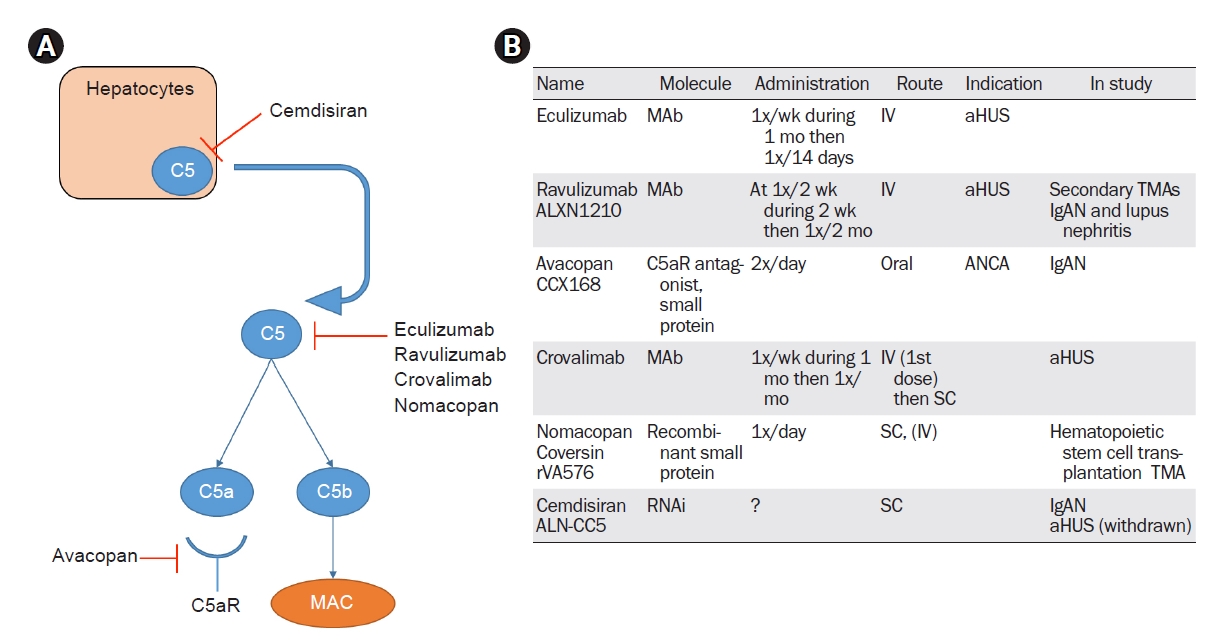
Anti-C5 therapies.
(A) A summary of the different mechanisms of inhibition of the terminal pathway. Cemdisiran, an RNA inhibitor, blocks the production of C5 in hepatocytes. Eculizumab, ravulizumab, crovalimab, and nomacopan inhibit the cleavage of C5 into C5a and C5b. Avacopan is an antagonist of the C5a receptor involved in inflammation pathways. (B) A summary of the different characteristics of anti-C5 therapies in kidney diseases.
aHUS, atypical hemolytic uremic syndrome; ANCA, antineutrophil cytoplasmic antibody; IgAN, immunoglobulin A nephropathy; IV, intravenous; MAb, monoclonal antibody; MAC, membrane attack complex; RNAi, ribonucleic acid inhibitor; SC, subcutaneous; TMA, thrombotic microangiopathy.
Atypical hemolytic uremic syndrome
For almost 10 years, anti-C5 therapy for aHUS has produced impressive results, with a significant decline in the number of patients on chronic dialysis after aHUS, a simultaneous increase in the number of patients with preserved renal function, and a similar increase in the number of patients with functioning grafts after transplantation for aHUS [8]. In 2021, new data became available regarding the safety profile, consequences of discontinuation, and benefits of ravulizumab, a new anti-C5 inhibitor.
Concerning the safety of eculizumab, a 5-year safety analysis from a registry cohort of 865 patients (535 adults and 330 children) treated with ≥1 dose of eculizumab for the indication of aHUS has been reported [9]. This group was compared to 456 aHUS patients that had never been treated (307 adults and 149 children). Meningococcal infection occurred in one child and two adults, which represents 0.11 and 0.17 events, respectively, per 100 patient-years. Of the three patients with invasive meningococcal diseases, two had not received antibiotic prophylaxis. Other patients were also at risk of serious infection (e.g., aspergillus infections or infections due to encapsulated bacteria such as Neisseria gonorrhea, Streptococcus pneumoniae, and Haemophilus influenzae), with a rate of 7.48 events per 100 patient-years in adults and 5.15 events per 100 patient-years in children. In comparison, the control group had 6.17 and 1.12 events, respectively, per 100 patient-years. Death occurred in 4.7% of treated adults and 1.8% of treated children. Infection remained the main cause of death, being responsible for 33% of cases. In the treated cohort, death was less frequent in the group of untreated adults (9.9%) but more frequent than in untreated children (0%). This difference could be explained by the hypothetical frailty of untreated adult patients and by less severe disease in the pediatric setting [9].
The significant information about invasive meningococcal infections has been confirmed in a registry study including PNH and aHUS patients [10] and those with neuromyelitis optica [11]. It was reported respectively as 0.25 and 0.54 events per 100 patient-years. Due to systematic vaccination against the different serotypes of meningoccocus, ACYW and B, and to the use of penicillin prophylaxis, the rate of infection has routinely decreased since 2010 and the mortality rate has remained very low.
In view of the risk of infection and the cost of treatment, eculizumab discontinuation must be discussed and weighed against the risk of aHUS recurrence. A recent prospective study [12] including 55 patients (both pediatric and adult) with a history of aHUS treated by eculizumab studied the outcome after drug discontinuation. Among these patients, 51% had a complement gene variant associated with aHUS. Of patients without genetic variants, only one experienced relapse. This unique case was reclassified after the study as congenital thrombotic thrombocytopenic purpura (ADAMTS13 activity at 5% and a pathogenic variant in the ADAMTS13 gene). Among patients who have a variant in complement genes, relapses were more frequent (12 of 28 patients, 42.9%). These relapses occurred mainly during or after an infection. Patients should, therefore, be carefully monitored, especially around infectious episodes. The rate of relapse could be overestimated because 16% of the patients had already experienced ≥1 relapse before inclusion [13]. Multivariate analysis showed an increased risk of relapse for the patients treated with eculizumab with a plasma-soluble C5b9 of ≥300 ng/mL at the start of the study. These relapses were treated with eculizumab, and 11 of 13 patients regained their baseline creatinine levels. During the whole study, eculizumab was not administered for a median of 24 months, and the cost savings were estimated at €32,000,000 [12].
Ravulizumab, another humanized monoclonal antibody that targets the same epitope on the C5 protein as eculizumab, has shown promising results in aHUS [14]. This drug was engineered from eculizumab to have a longer half-life, resulting in an infusion rate of every 8 weeks instead of every 2 weeks with eculizumab. This phase III study (ALXN1210-aHUS-311) showed that ravulizumab could induce a complete thrombotic microangiopathy (TMA) response in 53.6% of patients within 26 weeks. An improvement in renal function was observed in 68% of patients, and dialysis weaning was possible in 58% of patients on dialysis at baseline. This study was a single-arm trial and was not designed to compare ravulizumab and eculizumab. In the C10-004 study evaluating the effect of eculizumab in aHUS, the following results were obtained [2]: a complete TMA response was achieved in 56% of patients, any improvement of renal function in 54% of the patients and among those dialyzed at the baseline, 83% could be weaned of this technique. However, the difference in rates of dialysis weaning between ravulizumab and eculizumab (58% vs. 83%) raised concerns [15]. This could be explained by the different definitions and populations included in the two studies. Populations differed with the inclusion of 1) Asian centers with a significantly less-complete renal response and 2) fewer patients with a pathogenic variant in complement-related genes (57% in the eculizumab group vs. 31% in the ravulizumab group) [16]. The median time to achieve a complete TMA response was also increased among patients treated by ravulizumab (86 days vs. 57 days). Within the extension period of the ALXN1210-aHUS-311 study [16], four more patients attained complete TMA responses, increasing the treatment-response group to 61% of the total number of patients, and the renal response was long-lasting. The safety profile of ravulizumab is similar to eculizumab in the initial and extended studies but still requires confirmation in larger cohorts. Ravulizumab has also been studied in children and adolescents and appears safe and effective in a prospective uncontrolled study including 18 patients who have not previously received complement inhibitors [17].
The single-molecule crovalimab will be examined in a phase III study (COMMUTE-a and -p) for the indication of aHUS in adults or pediatric patients. Crovalimab is a long-acting C5 inhibitor that could be administered subcutaneously.
The conclusion is as follows.
• Anti-C5 therapies carry a risk of infectious complications in the real-life setting, but the risk of invasive meningococcal infection can be modulated with appropriate vaccinations and antibiotic prophylaxis.
• Eculizumab can be discontinued in aHUS patients, leading to a relapse rate of <5% in aHUS patients without a pathogenic variant in complement genes and with a relapse rate of approximately 50% in aHUS patients with a pathogenic variant. When a discontinuation is proposed, careful follow-up should occur, especially during or after an infectious event.
• Ravulizumab, another anti-C5 therapy, with a longer half-life, is effective in aHUS, but its noninferiority compared to eculizumab has not been established thus far in comparative trials.
Other thrombotic microangiopathies
Whereas the causal role of the alternative pathway is well described in aHUS, it is less clear in other forms of TMAs, e.g., Shiga toxin-associated hemolytic uremic syndrome (STEC-HUS) or secondary TMAs [18]. If the alternative pathway is not the primum movens of kidney lesions, then anti-C5 therapies would not be effective.
During the 2011 outbreak of STEC-HUS in northern Europe (mainly Germany), eculizumab was evaluated in two large retrospective studies and did not show efficacy for kidney outcomes or mortality [19,20]. Multiple case reports and case series have demonstrated a potential benefit of eculizumab in secondary TMAs, but all the reports are retrospective, lacking control groups, and the relative efficacy is subject to a publication bias [21,22].
Similar patient profiles are not shown in genetic studies of aHUS or other forms of TMA. Whereas a rare variant (allele frequency < 0.1%) in complement genes or anti-factor H antibodies are found in ~50% to 60% of patients with aHUS, it is only present in ~5% of STEC-HUS and secondary HUS cases [23,24]. In contrast, primary aHUS may be encountered after renal transplantation as a recurrence of the initial disease, during or after pregnancy, and during malignant hypertension; in these cases, rare variants are found in, respectively, 29%, 56%, and 51% of patients [25–27]. It is important to remember that genetic analyses in TMAs are complex to interpret and require the expertise of a specialized laboratory. In addition, since results are not rapidly available, therapeutic management cannot be delayed until genetic analysis is performed.
To circumvent this problem, a functional test like ex vivo analysis has been developed by different study groups [28]. It consists of incubating patient serum in vitro on cultured endothelial cells (mainly immortalized human dermal microvascular endothelial [HMEC-1] cells) and quantifying C5b9 deposits using confocal microscopy. Promising results have been published regarding aHUS, with high C5b9 deposition noted in the acute phase of aHUS, which decreases after remission. Interestingly, when HMEC-1 cells are activated by adenosine diphosphate, patients in remission or even those who are asymptomatic carriers of pathogenic variants in complement genes showed an increased deposition of C5b9 [28]. This test has been evaluated in malignant hypertension patients and showed that, in 26 patients, 18 had a massive deposition of C5b9; further, this subgroup included all patients with a pathogenic variant in complement genes (9 of 18, 50%) [29]. This test has also been evaluated in other secondary TMAs, and ex vivo complement activation was found in a proportion of patients varying between 59% and 100% [30–32], with some indirect evidence of eculizumab efficacy. It should be stressed that this functional test is difficult to perform and to reproduce in nonspecialized laboratories, so a robust test allowing rapid identification of complement-mediated aHUS has yet to be developed. In addition, strong clinical evidence of the efficacy of anti-C5 drugs in secondary HUS is lacking.
This year, Alexion Pharmaceuticals (Boston, MA, USA) launched a randomized, placebo-controlled clinical trial to evaluate the efficacy of ravulizumab in some secondary TMAs (NCT04743804) but excluding, for example, patients who are pregnant or who have cancer. AKARI Therapeutics (New York, NY, USA) will also evaluate nomacopan in pediatric hematopoietic stem-cell transplant–associated TMA during a phase III, open-label, uncontrolled trial (NCT04784455). These studies will increase our knowledge of C5 inhibition in secondary TMAs.
C3 glomerulopathy
C3G, including dense deposit disease (DDD) and C3 glomerulonephritis (C3GN), is another disease implicating a dysregulation of the alternative pathway. Some patients carry a rare variant in complement-related genes, and others have auto-antibodies potentializing C3 and/or C5 convertase, like C3 and C5 nephritic factors [33]. This disease could theoretically benefit from C5 inhibition.
The incidence of this rare disease is difficult to estimate and may vary between 1 and 3 cases, respectively, per 1,000,000 people [33] and, since classification in C3G is not yet well defined, it is problematic to perform randomized controlled trials. To date, two retrospective studies [34,35] and two prospective uncontrolled trials have attempted evaluation of the efficacy of eculizumab in this indication [36,37]. The first study in 2012 was a proof-of-concept study evaluating the efficacy of eculizumab in six patients (three with DDD and three with C3GN) with a protocol biopsy after 1 year of treatment. As a potential predictive marker of the response to treatment, the authors suggested an elevated soluble C5b9 level in the serum at the initiation of eculizumab. This elevated sC5b9 concerned only three patients of the six; two of whom had a good clinical response [36]. The following two trials were retrospective series, the first reporting seven patients (five with C3GN and two with DDD) [35] and the second assessing all 26 patients with C3G treated by eculizumab from a French registry [34]. Welte et al. [35] reported favorable outcomes (improvement or stabilization of the renal function) in five of their seven patients. The French registry study reported an overall clinical response in 23% and a partial clinical response in another 23% of patients, respectively. Factors associated with the overall clinical response included a greater proportion of rapidly progressive glomerulonephritis, with a low estimated glomerular filtration rate at eculizumab initiation and the presence of cellular crescents on biopsies. No statistical difference was observed in the serum level of soluble C5b9 in this study [36].
A prospective off-on-off-on clinical trial without a control group included six patients with immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and four patients with C3G. All included patients had a serum sC5b9 levels of >1,000 ng/mL and 24-hour proteinuria levels of >3.5 g. Only three of the 10 patients achieved a response to treatment (two partial and one complete remission), defined by a reduction of 50% of proteinuria and the excretion of <3.5 g/24 hr for partial and <0.3 g/24 hr for complete remission, respectively. This study is also interesting in that it shows that eculizumab was effective in dramatically reducing the level of sC5b9 in all patients, with only a few clinical responses [37].
It remains to be determined whether the proximal part of the complement cascade, e.g., C3, C3a, and C3b, actually plays no major part in this disease. To answer this question, there are plans to study three molecules in C3G: iptacopan, a factor B inhibitor; danicopan, a factor D inhibitor; and narsoplimab, a lectin pathway inhibitor.
In conclusion, eculizumab could be of benefit for some patients, such as those with crescentic forms of C3G. However, these observations are based on a low number of patients and should not be considered as recommendations.
Antineutrophil cytoplasmic antibody vasculitis
The pathogenesis of ANCA-associated vasculitis is known to involve the complement system. Neutrophils play a central role in this disease and, after being primed by cytokines, they are responsible for endothelial lesions. At the same time, neutrophils induce the release of properdin and factor B, which are crucial for alternative pathway activation. The alternative pathway results in the generation of C5a, which amplifies the inflammatory response by recruiting and priming other neutrophils [38,39].
In an experimental model of adeno-associated viruses in mice, it was found that C5a and C5aR were key players in the vascular lesions [40]. Thereafter, avacopan, an oral antagonist of C5a receptors, was evaluated in two phase II studies (CLEAR and CLASSIC). The CLEAR study showed noninferiority of avacopan for the clinical response and the safety profile in this pathology [41]. Despite the trial being designated a noninferiority study, there were signs of greater efficacy for avacopan when considering the Birmingham Vasculitis Activity Score (BVAS). The study included 67 patients divided into three study groups: high-dose prednisone (60 mg daily), lower-dose prednisone (20 mg daily) + 30 mg of avacopan twice daily, and 30 mg of avacopan twice daily alone. The CLASSIC study evaluated two different doses of avacopan (10 or 30 mg twice daily) in combination with standard of care (SOC) vs. SOC alone [42]. It also showed a good safety profile, and a higher dose of avacopan seemed to reduce the time to remission.
Recently, there have been reports from a phase III study (ADVOCATE) [43] designed to compare corticosteroids vs. avacopan, both with cyclophosphamide or rituximab. The corticosteroid group received a tapering dose of prednisone until day 140, adapted to each patient’s weight and age. This corresponded with a starting dose of 60 mg of prednisone for an adult weighing ≥55 kg. The avacopan group received 30 mg twice daily during the 52 weeks of the study period. Both groups also received a placebo and rituximab for 4 weeks or cyclophosphamide for 12 weeks, followed by azathioprine at week 15. The use of glucocorticoids was authorized during the screening period, and 75% of the patients received a prednisone-equivalent dose of 46.7 ± 53.2 mg/day in the avacopan group.
No differences were observed between the two groups regarding remission at week 26 (72.3% vs. 70.1%, respectively, for avacopan and corticosteroids), but avacopan was superior for achieving sustained remission at week 52 (65.7% vs. 54.9%, respectively). This is an important breakthrough for anticomplement therapies because it could reduce the infectious morbidity associated with this disease (any infection in 68.1% vs. 75.6%, respectively, and any serious opportunistic infection in 3.6% vs. 6.7%, respectively; for avacopan and prednisone).
However, the tapering regimen of the prednisone group is questionable with interruption of the glucocorticoids at week 20. Interruption was faster than other studies in ANCA vasculitis as exemplified by the PEXIVAS study, which conserved glucocorticoids at least until week 52, even in the reduced-dose regimen [44]. A meta-analysis also showed that longer courses of glucocorticoids were associated with fewer relapses [45]. This rapid weaning off of glucocorticoids could increase the rate of relapses at week 52 when, at the same time, continuous treatment was available in the avacopan group. On the other hand, it could underestimate the infectious risk of a longer regimen of glucocorticoids.
In conclusion, avacopan could in part replace glucocorticoids in ANCA vasculitis to reduce the well-known side effects of cortisone, with the limits as previously described—namely, an infusion of glucocorticoids in avacopan patients and a rapid weaning of glucocorticoids in the other group.
Immunoglobulin A nephropathy and lupus nephritis
IgA nephropathy (IgAN) is a very common cause of glomerulonephritis worldwide and consequently of chronic kidney disease. The complement system seems to be implicated in the disease, with genome-wide significance studies identifying the CFHR gene family as a susceptibility locus, with opposing effects noted for individual CFHR genes; for example, homozygous CFHR1/CFHR3 deficiency is protective, whereas enhanced FHR5 plasma levels is an independent risk factor [46]. Frequent mesangial depositions of IgA with C3 and C5b9 are observed, and there is even a possible correlation between the intensity of C5b9 deposition and disease severity [7].
Experimental studies in an IgAN mouse model also demonstrated that the knockout strains for C3aR or C5aR had lower IgA deposition in the mesangium. The use of C3aR and C5aR antagonists reduced in vitro IgA-induced cell proliferation and production of interleukin-6 and MCP-1, which are involved in inflammation pathways [47]. Few case reports report a potential positive effect of C5 inhibition [48,49], but no randomized control trials have yet been published.
A phase II study was launched in 2015 for the evaluation of the C5aR antagonist avacopan in IgAN (NCT02384317), but no results are available yet. A study of cemdisiran (NCT03841448) is planned for this indication. Cemdisiran is an RNA inhibitor that specifically targets the liver and blocks hepatic production of C5. The efficacy and safety of ravulizumab (NCT04564339) will be assessed in patients with proliferative lupus nephritis or IgAN in a phase II clinical trial.
Lupus nephritis is a frequent clinical manifestation of systemic lupus erythematosus. This disease is associated with deposition of the immune complex with the complement component of the classical pathway (“full house pattern”) in the kidneys. Some mouse models have shown that it could be beneficial to target C3aR or C5aR to reduce kidney inflammation [50].
In conclusion, other glomerular diseases like IgAN or lupus nephritis could benefit from anticomplement therapies, but there is no actual evidence of efficacy available so far.
Conclusion
In recent years, there have been remarkable results from C5 inhibition in primary aHUS, including those triggered by pregnancy or after kidney transplantation, and there have been interesting outcomes from the C5aR antagonist in ANCA vasculitis. The efficacy of these drugs needs to be studied in larger clinical trials and to be evaluated in the context of secondary TMA. Several trials are in progress at present. New, long-acting C5 inhibitors, such as ravulizumab or crovalimab, may alleviate the burden of chronic treatments. Subcutaneous and oral forms of complement inhibitors may also improve treatment tolerance and compliance. Importantly, C5 blockade requires careful monitoring as well as antimeningococcal vaccination and prophylactic antibiotherapy to reduce the infectious risk inherent with these drugs.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Authors’ contributions
Conceptualization, Formal analysis: AW
Writing - original draft preparation: AW
Writing - review & editing: ER
All authors read and approved the final manuscript.