A calpain inhibitor protects against fractalkine production in lipopolysaccharide-treated endothelial cells
Article information
Abstract
Background
Fractalkine (CX3CL1) is a chemokine with a unique CX3C motif and is produced by endothelial cells stimulated with lipopolysaccharide (LPS), tumor necrosis factor (TNF)-α, interleukin (IL)-1, and interferon-γ. There have been several reports that the caspase/calpain system is activated in endotoxemia, which leads to cellular apoptosis and acute inflammatory processes. We aimed to determine the role of the caspase/calpain system in cell viability and regulation of fractalkine production in LPS-treated endothelial cells.
Methods
Human umbilical vein endothelial cells (HUVECs) were stimulated with 0.01–100 μg/mL of LPS to determine cell viability. The changes of CX3CL1 expression were compared in control, LPS (1 μg/mL)-, IL-1α (1 μg/mL)-, and IL-1β (1 μg/mL)-treated HUVECs. Cell viability and CX3CL1 production were compared with 50 μM of inhibitors of caspase-1, caspase-3, caspase-9, and calpain in LPS-treated HUVECs.
Results
Cell viability was significantly decreased from 1 to 100 μg/mL of LPS. Cell viability was significantly restored with inhibitors of caspase-1, caspase-3, caspase-9, and calpain in LPS-treated HUVECs. The expression of CX3CL1 was highest in IL-1β-treated HUVECs. CX3CL1 production was highly inhibited with a calpain inhibitor and significantly decreased with the individual inhibitors of caspase-1, caspase-3, and caspase-9.
Conclusion
The caspase/calpain system is an important modulator of cell viability and CX3CL1 production in LPS-treated endothelial cells.
Introduction
Sepsis is caused mainly by an exaggerated systemic response to endotoxemia, induced by gram-negative bacteria and their characteristic cell wall component, lipopolysaccharide (LPS) [1]. LPS is known to cause acute increases in the number and density of adhesion molecules in the vascular endothelium [2]. As an inflammatory mediator, LPS can cause damage to the vascular endothelium that can, in turn, lead to capillary leak syndrome, dilation of blood vessels, and a decrease in cardiac function, all contributing to septic shock [3].
On the injured endothelium, fractalkine, also known as CX3CL1, functions as both a potent chemoattractant and an adhesion molecule for the recruitment and migration of fractalkine receptor (CX3CR1)-expressing circulating inflammatory cells into the sites of injury and inflammation [4]. CX3CL1 is a major chemoattractant for macrophages that express CX3CR1 [5,6]. CX3CL1 has been implicated as a mediator in several inflammatory conditions, including atherosclerosis, atopic dermatitis, multiple sclerosis, and Crohn’s disease [7–9]. Recent studies have suggested that CX3CL1 plays a role in the pathogenesis of sepsis. Mice with polymicrobial abdominal sepsis caused by cecal ligation and puncture (CLP) have elevated CX3CL1 levels in their peritoneal lavage fluid and serum [10–12]. Sung et al [13] reported that CX3CL1 was expressed in arterial endothelial cells in an LPS-induced endotoxemic model in rats.
Caspases are proteases that are central players in apoptosis induction and participate in the promotion of apoptotic cell death with the caspase-8, -9, and -3 cascade. The calpains are a family of calcium-dependent proteases that are involved predominantly in intracellular signaling, cytoskeletal stability, apoptosis, and acute inflammatory processes. There have been several reports that the caspase/calpain system is activated in endotoxemia that leads to cellular apoptosis and acute inflammatory process [14–16]. Guo et al [17] recently reported that acute renal failure from endotoxemia was dependent on caspase activation; in the same study, macrophage inflammatory protein-2 (MIP-2, CXCL2) secretion was decreased with caspase inhibition in LPS-treated endothelial cells [17]. Based on these findings, we aimed to determine the role of the caspase/calpain system in cell viability and regulation of fractalkine production in LPS-treated endothelial cells.
Methods
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (C2519A-lonza; Walkersville, MD, USA) and used in the experiments. The cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 5% heat-inactivated fetal calf serum, 1% penicillin streptomycin, 4 mM L-glutamine, 4.5 g/L glucose, and 1.5 g/L sodium bicarbonate. Cells were kept in a humidified incubator with 5% CO2-95% air at 37°C, and the media were refreshed two to three times weekly. The cells were maintained in six-well culture plates, and the passages were performed by trypsinization. The experiments were performed when the plates became at least 80% confluent.
LPS, interleukin (IL) 1α, IL-1β, and media were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-fractalkine (CX3CL1) antibody was purchased from Torrey Pines Biolabs (Secaucus, NJ, USA). Calpain inhibitor (BML-PI116-0005) and caspase-1 inhibitor (ZYVAD[OMe]-FMK) were purchased from Enzo Life Sciences (Farmingdale, NY, USA). Caspase-3 inhibitor (Ac-DEVD-CHO) and caspase-9 inhibitor (Z-LEHD-FMK) were purchased from BD Biosciences (San Jose, CA, USA).
Treatment of LPS and proinflammatory cytokines
HUVECs were seeded in six-well cell culture plates. The following day, the cells were washed twice with 200 μL of cold phosphate-buffered saline (PBS) and stimulated with 0.01, 0.1, 1, 10, or 100 μg/mL of LPS for 24 hours in culture medium in order to investigate cell viability and the expression of CX3CL1. In separate experiments, the cells were similarly treated with 1 μg/mL concentrations of IL-1α and IL-1β for 24 hours in order to elucidate the roles of IL-1α and IL-1β in CX3CL1 expression. The control cells were treated with vehicle (0.1% DMEM).
Cell viability test
Cell viability was determined using the CellCountEZTM Cell Survival Assay Kit (Rockland Immunochemicals, Limerrick, PA, USA). Cells were washed twice with 200 μL of cold PBS and incubated in 20% 3-(4, 5-dimeth-ylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) in FBS-free Human Endothelial-Serum Free Media (HE-SFM) for 1 hour at 37°C with 5% CO2. The amount of formazan converted by viable cells was determined by measuring absorbance at 490 nm on a 96-well microplate reader after subtracting the absorbance at 650 nm [18].
Caspases and calpain inhibition
To determine the roles of caspases and calpain in LPS-treated HUVECs, cells were incubated with either 50 μM calpain inhibitor (BML-PI116-0005), caspase-1 inhibitor (ZYVAD[OMe]-FMK), caspase-3 inhibitor (Ac-DEVD-CHO), or caspase-9 inhibitor (Z-LEHD-FMK) 2 hours before the LPS treatment.
Real-time polymerase chain reaction (PCR) of CX3CL1
For the PCR experiments, HUVECs were cultured in 12-well plates, and total RNA was extracted using an RNeasy kit (Qiagen, Hilden, Germany). Total RNA was reverse-transcribed using a cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Briefly, the reaction was performed in a final volume of 20 μL that included 100 mM dNTP, random primers, MultiScribe™ Reverse Transcriptase, RNase inhibitor, and 1 μg total RNA. The reaction mixtures were incubated at 25°C for 10 minutes, then at 37°C for 120 minutes, and finally at 85°C for 5 seconds. Real-time PCR was performed using a StepOne PCR System (Applied Biosystems) in triplicate and in a final volume of 20 μL that included TaqMan gene expression master mix with an optimized concentration of each primer, 250 nM TaqMan probe, and 2.0 μL cDNA reaction mixture. The reaction mixtures were preheated at 95°C for 10 minutes to activate the enzyme and then subjected to 40 cycles of melting at 95°C for 15 seconds and annealing/extension at 60°C for 1 minute. The real-time PCR efficiencies were approximately 100%. The assay-on-demand gene expression products (Applied Biosystems) were used to evaluate the mRNA expression levels of CX3CL1 (No. Hs00171086_m1) and 18S rRNA (No. Hs99999901_s1). The primers used for fractalkine were forward 5′-TGT GTC TCT TGG TCC TGC TG-3′ and reverse 5′-GAT CAG AAC CCA GCC TTC AG-3′. The 18S rRNA was used as an internal control. For each sample, the mRNA levels were normalized against the 18S rRNA levels, and the ratios of normalized mRNA to the untreated control sample were determined using the comparative Ct method [19].
Immunoblotting for CX3CL1
HUVECs were seeded in six-well collagen-coated cell culture plates and stimulated as indicated. Following stimulation, culture supernatants were collected and centrifuged at 4,000 rpm for 5 minutes. The supernatants were treated with 0.25% sodium dodecyl sulfate (SDS; final), 1 mM DTT (final), and a protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) and boiled for 5 minutes. The boiled mixture was cooled to room temperature and passed through AmiconUltra-4 Centrifugal Filter Devices (molecular weight cutoff: 50,000). Proteins in the filtrate were concentrated with trichloroacetic acid precipitation. The precipitate was dissolved in 20 μL of SDS-sample buffer and subjected to 15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), followed by Western blot analysis of CX3CL1 content as previously described [20].
Statistical analysis
Each experiment was performed at least three times. All values are expressed as the mean ± standard deviation. Statistical analyses were performed using unpaired, two-tailed Student t-tests. The data were corrected by Dunn’s method. A P value < 0.05 was considered statistically significant.
Statistical analyses were performed using SPSS software ver. 14.0 (SPSS Inc., Chicago, IL, USA).
Results
Endothelial cell viability after LPS treatment
The cell viability after treatment with LPS at 1, 10, or 100 μg/mL was 0.78, 0.75, or 0.76 fold, respectively, compared to that of the control. However, endothelial cell viability after treatment with LPS at 0.01 or 0.1 μg/mL was not significantly different from that of the control (Fig. 1).

Endothelial cell viability after lipopolysaccharide (LPS) treatment
The cell viability of human umbilical vein endothelial cells (HUVECs) was significantly decreased after treatment with LPS at 1, 10, or 100 μg/mL compared to that of HUVECs treated with LPS at 0 (control), 0.01, or 0.1 μg/mL.
*P < 0.05 vs. 0, 0.01, 0.1 μg/mL of LPS treatment. Bars represent the mean ± standard deviation from three experiments.
Endothelial cell viability after treatment with inhibitor of caspase-1, -3, -9, or calpain
The cell viability of LPS-treated HUVECs was significantly restored after treatment with an inhibitor of cas-pase-1, caspase-3, caspase-9, or calpain (Fig. 2). In particular, cell viability was most restored with the caspase-3 inhibitor treatment. There was no significant difference in restored cell viability of LPS-treated HUVECs among inhibitors of calpain, caspase-1, and caspase-9 (Fig. 2).
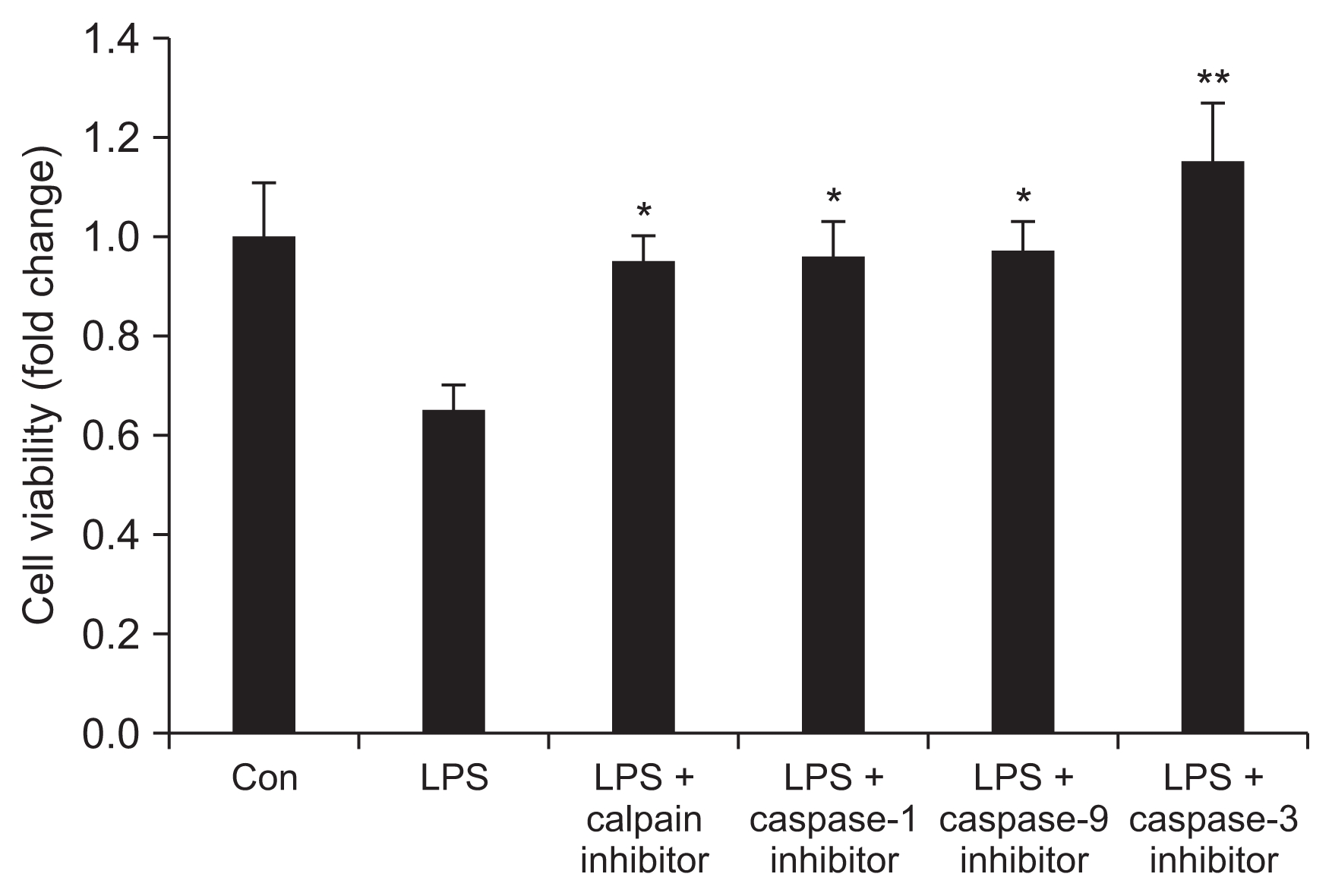
Endothelial cell viability after treatment with inhibitors of calpain, caspase-1, -9, and -3
Human umbilical vein endothelial cells were incubated with inhibitors of calpain, caspase-1, -9, or -3 for 2 hours before lipopolysaccharide (LPS) treatment.
Con, control; LPS, LPS 1 μg/mL; calpain inhibitor, 50 μM of calpain inhibitor; caspase-1 inhibitor, 50 μM of caspase-1 inhibitor; cas-pase-9 inhibitor, 50 μM of caspase-9 inhibitor; caspase-3 inhibitor, 50 μM of caspase-3 inhibitor.
*P < 0.05 vs. LPS; **P < 0.05 vs. LPS, caspase-1, caspase-9, calpain. Bars represent the mean ± standard deviation from three experiments.
CX3CL1 mRNA expression in HUVECs after LPS treatment
The mRNA expression of CX3CL1 in LPS-treated HU-VECs significantly increased with as little as 0.1 μg/mL of LPS treatment and showed a dose-dependent increment with additional LPS (1–100 μg/mL of LPS) (Fig. 3).
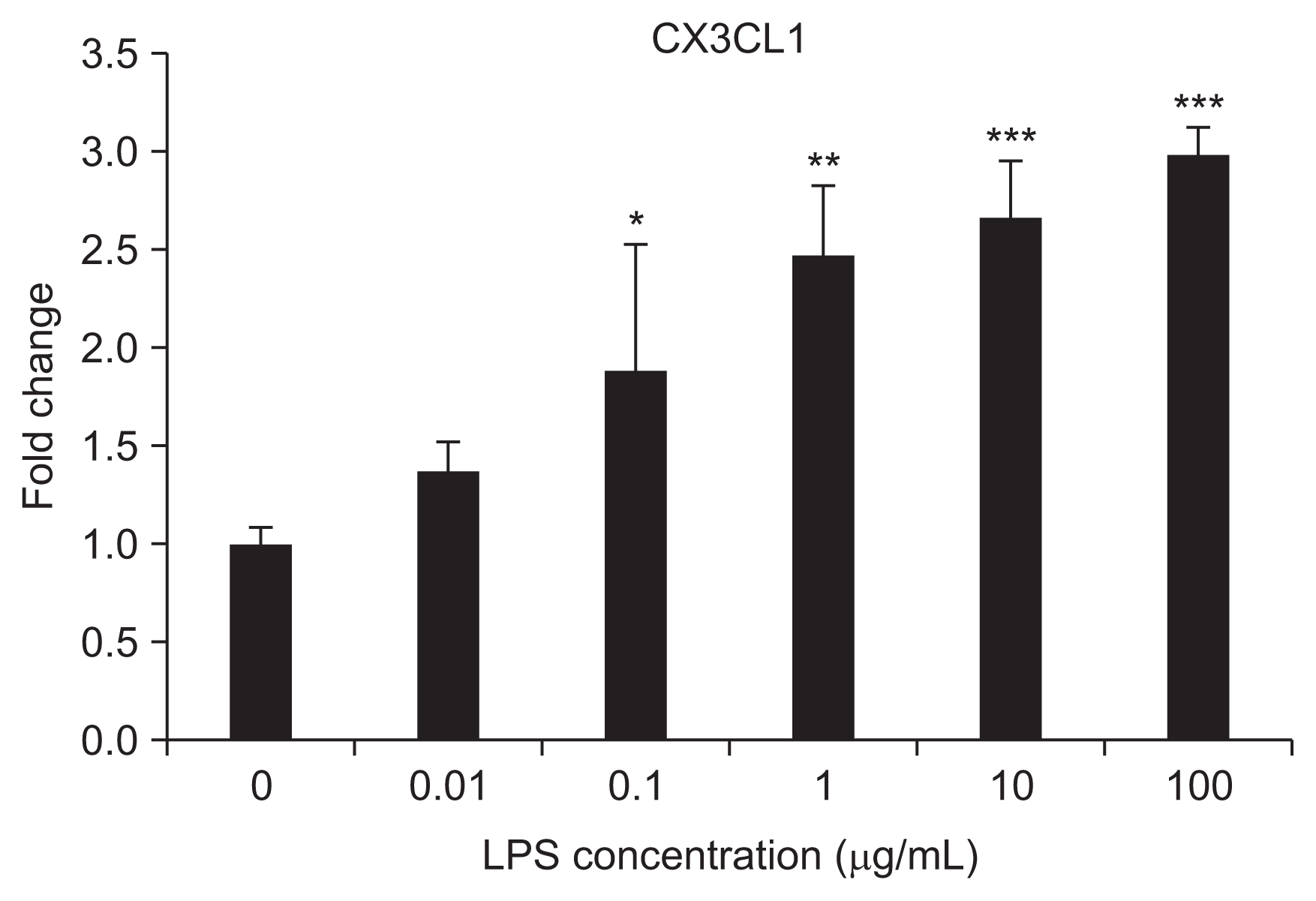
CX3CL1 mRNA expression in human umbilical vein endothelial cells after lipopolysaccharide (LPS; 0.01–100 μg/mL) treatment
*P < 0.05 vs. 0 (control), 0.01 μg/mL; **P < 0.05 vs. 0, 0.01, 0.1; ***P < 0.05 vs. 0, 0.01, 0.1, 1 μg/mL. Bars represent the mean ± standard deviation from three separate experiments.
CX3CL1 mRNA expression in HUVECs after proinflammatory cytokine treatment
The mRNA expression of CX3CL1 was highest in IL-1β-treated HUVECs (80 fold) compared to control cells. Treatment-led increases in the CX3CL1 message were also seen with LPS (3-fold change) and IL-1α (30 fold) with the HUVECs (Fig. 4).
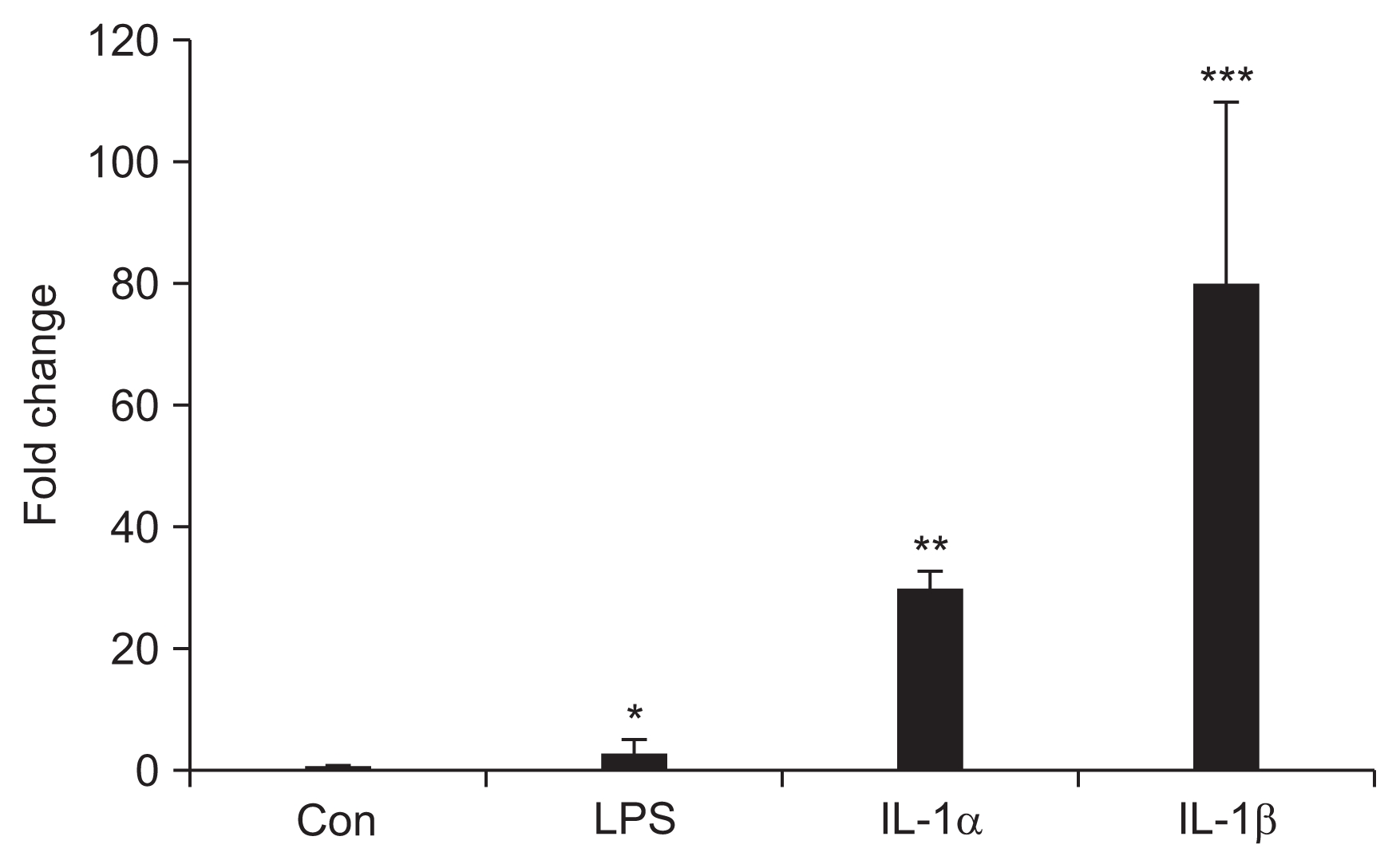
CX3CL1 mRNA expression in human umbilical vein endothelial cells after proinflammatory cytokines treatment
Con, control; LPS, 1 μg/mL of lipopolysaccharide; IL-1α, 1 μg/mL of interleukin-1α; IL-1β, 1 μg/ml of IL-1β.
*P < 0.05 vs. Con; **P < 0.05 vs. Con, LPS; ***P < 0.05 vs. Con, LPS, IL-1α. Bars represent the mean ± standard deviation from three separate experiments.
CX3CL1 mRNA expression in HUVECs with inhibitors of caspase-1, -3, -9, and calpain
The mRNA expression of CX3CL1 was significantly affected by treatment of the HUVECs with inhibitors of calpain, caspase-1, -3, and -9. In particular, the expression of CX3CL1 was most inhibited with calpain inhibitor treatment (Fig. 5).

CX3CL1 mRNA expression in human umbilical vein endothelial cells (HUVECs) after treatment with inhibitors of calpain, caspase-1, -9, and -3
HUVECs were incubated with inhibitors of calpain, caspase-1, -9, and -3 for 2 hours before lipopolysaccharide (LPS) treatment.
Con, control; LPS, LPS 1 μg/mL; calpain inhibitor, 50 μM of calpain inhibitor; caspase-1 inhibitor, 50 μM of caspase-1 inhibitor; caspase-9 inhibitor, 50 μM of caspase-9 inhibitor; caspase-3 inhibitor, 50μM of caspase-3 inhibitor.
*P < 0.05 vs. LPS; **P < 0.05 vs. LPS, caspase-1, caspase-9, caspase-3. Bars represent the mean ± standard deviation from three separate experiments.
CX3CL1 immunoblotting in endothelial cell
Starting from 0.1 μg/mL of LPS, the protein level of CX-3CL1 in LPS-treated HUVECs was significantly increased (Fig. 6A). The production of CX3CL1 was highest in IL-1β-treated HUVECs compared to the control, LPS-, and IL-1α-treated HUVECs (Fig. 6B). The protein production of CX3CL1 was significantly decreased with inhibitors of calpain, caspase-1, -3, and -9, and this effect was particularly notable with the calpain inhibitor (Fig. 6C).
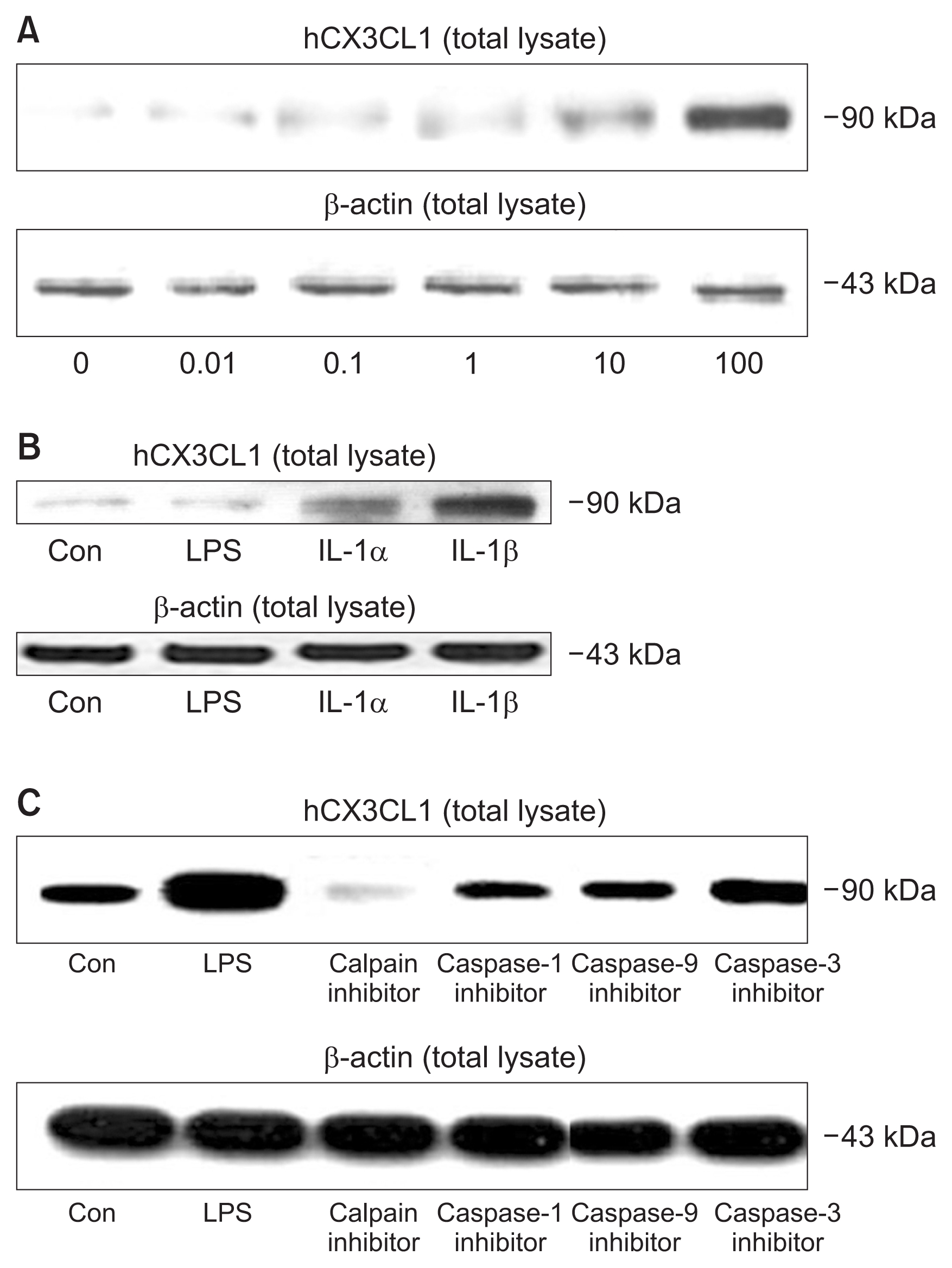
(A) CX3CL1 immunoblot from human umbilical vein endothelial cell (HUVEC) lysates after lipopolysaccharide (LPS; 0.01–100 μg/mL) treatment. Representative immunoblot of at least three separate experiments is shown. (B) CX3CL1 immunoblotting expression in HUVECs after proinflammatory cytokines treatment. (C) CX3CL1 immunoblotting in HUVECs after treatment with inhibitors of calpain, caspase-1, -9, and -3.
Con, control; LPS, 1 μg/mL of LPS; IL-1α, 1 μg/mL of interleukin (IL)-1α; IL-1β, 1 μg/mL of IL-1β; calpain inhibitor, 50 μM of calpain inhibitor; caspase-1 inhibitor, 50 μM of caspase-1 inhibitor; caspase-9 inhibitor, 50 μM of caspase-9 inhibitor; caspase-3 inhibitor, 50 μM of caspase-3 inhibitor. Representative immunoblot of at least three separate experiments is shown.
Discussion
The main findings of our study were as follows: 1) Cell viability was significantly restored with inhibitors of cal-pain, caspase-1, caspase-3, and caspase-9 in LPS-treated HUVECs; 2) The mRNA expression of CX3CL1 was highest in IL-1β–treated HUVECs; and 3) mRNA expression and production of CX3CL1 were highly inhibited by a calpain inhibitor.
During the regulation of endothelial cell apoptosis during sepsis, there are two major pathways involved in the initiation of apoptotic death (the activation of pro-caspase-8 in the death receptor pathway or by caspase-2 and caspase-9 in the mitochondrial apoptosis pathway), both of which ultimately converge to activate executioner caspase-3 [21]. The death receptor pathway involves activation of members of the tumor necrosis factor (TNF) receptor family with an intracellular death domain including Fas, TNF-R1, DR3, and the receptors for TRAIL. After ligand binding to death receptors, the death domain recruits the adaptor protein FADD. FADD can then recruit pro-caspase-8 to the “death-inducing signal complex,” thereby causing its activation. Caspase-8 then activates caspase-3. LPS is well known as a death ligand. In addition, there have been many reports that caspase-8 inhibition prevents significant appearance of TUNEL-positive aortic endothelial cells [21]. This indicates that the death receptor pathway is mainly involved in LPS-induced endothelial cell apoptosis. The second apoptotic death pathway is a mitochondrial-medicated pathway. Following the mitochondrial release of apoptogenic compounds such as cytochrome c, caspase-9 activation occurs through the oligomerization mediator APAF-1, resulting in amplification of the death receptor-medicated pathway [21]. LPS activates caspase-1 in cultured endothelial cells. There is evidence that caspase-1, formerly termed the IL-1–converting enzyme, can mediate apoptotic and necrotic cell death [22,23]. In our results, cell viability was significantly restored by inhibitors of caspase-1, cas-pase-3, and caspase-9 in LPS-treated endothelial cells. This result suggests that caspase-1 and the mitochondrial cell death pathway might be involved in LPS-induced endothelial cell death. The studies of Choi et al [24] and Munshi et al [25] support our data.
Calpain is predominantly involved in intracellular signaling, cytoskeletal stability, necrosis, and apoptosis [26]. Calpain can cleave caspases-7, -8, and -9, resulting in their inactivation [3]. On the other hand, calpain can also cleave procaspase-12 to generate an active caspase and cleave the anti-apoptotic Bcl-XL molecule into a proapoptotic molecule [3]. Calpain can act a negative or a positive regulator of cell death pathways under different circumstances. Therefore, we investigated whether calpain was a mediator of LPS-induced endothelial cell death. In our study, we detected a significant increase in cell viability using calpain inhibitor. These results imply that calpain is a mediator of LPS-induced endothelial cell death.
Our data demonstrated a dose-dependent significant increase in CX3CL1 expression in LPS-treated endothelial cells, supporting a role for CX3CL1 as an important mediator in the pathogenesis of sepsis. Treatment of HUVECs with a variety of cytokines and LPS up-regulated CX3CL1 mRNA expression with an order of potency of IL-1β >> IL-1α >> LPS > control. The expression of CX3CL1 was highest in IL-1β–treated HUVECs compared to the control, LPS-, and IL-1α–treated HUVECs. This implies that IL-1β is the most potent stimulator in the endothelial expression of CX3CL1 compared to LPS or IL-1α. Brown et al [27] demonstrated that IL-1α was the most potent inducer of MCP-1 generation, whereas TNF-α induced maximal IL-8 secretion. These data suggest that the regulation of individual chemokine expression occurs through significantly different mechanisms. Such differences in the expression of specific chemokines can explain the specific appearance of various leukocytes at sites of inflammation and injury.
TNF-α, IL-1, and LPS initiate similar pathways in terms of activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) [28]. In rat aortic endothelial cells, the induction of CX3CL1 by IL-1β, TNF-α, and LPS is reported to be dependent on activation of NF-κB [29]. Calpains have been shown to degrade the inhibitor I-kB and thus induce the nuclear translocation of NF-kB [30]. There was a report that TCR-induced, PKC-θ-mediated NF-κB activation was regulated by the caspase-8, -9, -3 cascade [31]. Taken together, these findings suggest the possibility that the calpain and caspase system is a regulator of CX3CL1 expression in LPS-treated endothelial cells. Expectedly, our findings indicated that the expression of CX3CL1 was highly down-modulated by calpain inhibitors and significantly decreased with inhibitors of caspase-1, caspase-3, and caspase-9. Two studies reported that epigallocatechin-3-O-gallate (a major poly-phenol in green tea) and genistein (a major isoflavone in soy and red clover) suppressed TNF-α-induced fractal-kine mRNA and protein expression in primary cultured HUVECs through regulation of the NF-κB signaling pathway [32,33]. These studies led us to explore the possibility of calpain or caspase affecting fractalkine expression via NF-κB inactivation. So, elucidation of mechanisms in the calpain/caspase system affecting fractalkine expression merits further study.
In summary, cell viability was significantly restored with inhibitors of calpain and individual caspases, and the mRNA expression and production of CX3CL1 were highly inhibited with a calpain inhibitor in LPS-treated HUVECs. This implies that the caspase and calpain system is an important modulator of cell viability and CX-3CL1 production in LPS-treated endothelial cells.
Acknowledgments
This study was financially supported by Myongji Hospital Research Grant, 2013.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.