


Introduction
Diabetic nephropathy is one of the major complications of diabetes mellitus and currently the leading cause of end-stage renal disease worldwide [1]. Early clinical manifestations of diabetic nephropathy include hyperfiltration and microalbuminuria, which could progress to overt proteinuria and advanced renal injury. Accompanying morphological and ultrastructural changes include enlargement of glomeruli, mesangial expansion, thickening of the glomerular basement membrane, and effacement, denudation, and loss of podocytes [1]. Accompanying biochemical alterations with pathological changes lead to increases in the glomerular permeability as a result of the impaired glomerular filtration structure. These changes would be caused by hyperglycemia, glycated proteins, or irreversible advanced glycosylation end products (AGEs) via various mechanisms, including biochemical pathways, signaling, cytokines, and oxidative stress [2], [3].
AMP (adenosine monophosphate)-activated protein kinase (AMPK) is a ubiquitously expressed heterotrimeric kinase highly conserved from yeast to plants and animals that plays a key role in the regulation of energy homeostasis by coordinating multiple metabolic pathways [4], [5], [6]. AMPK is composed of a catalytic ╬▒ subunit and regulatory ╬▓ and ╬│ subunits, each of which is encoded by two or three distinct genes (╬▒1, 2; ╬▓1, 2; ╬│1, 2, 3) [4], [5], [6]. The kinase is activated by an elevated AMP/adenosine triphosphate (ATP) ratio due to cellular and environmental stress, such as heat shock, hypoxia, and ischemia [4], [5], [6].During energy stress as the level of ATP begins to fall, there is a marked increase in the cellular concentrations of AMP [4], [5], [6], [7]. This increase in AMP leads to the activation of AMPK via multiple mechanisms [7]. Once activated, AMPK acts to restore energy homeostasis by phosphorylating multiple substrates that act both to stimulate energy production and minimize energy consumption. The relationship between AMPK activation and the beneficial metabolic effects provides the rationale for the development of new therapeutic strategies for diabetic vascular diseases [8]. Thus, pharmacological AMPK activation may, through signaling and metabolic and genetic expression effects, reduce the risk of diabetic and metabolic complications.
Over the past decade, the biology and biochemistry of the AMPK pathway have been studied intensively in various organs, such as the liver, skeletal muscle, and heart. Although AMPK is abundantly expressed in the kidney [9], [10], its role in renal physiology and disease is less understood compared with that in other organs [11], [12]. In recent years, although interest in AMPK in the kidney has intensified, only a few studies have been conducted on the role of AMPKs in podocytes. We hypothesized that diabetic conditions would induce changes in AMPK in podocytes and that therapeutic modulation of AMPK would be a potential target in the treatment of diabetic podocytopathy.
Methods
Preparation of rat diabetic renal tissue
All animal experimental procedures were performed according to the guidelines for the care and use of animals established by Yeungnam University, Gyeongsan, Korea. Diabetes was induced by an intravenous injection of streptozotocin (STZ; Sigma Chemical Co., St. Louis, MO, USA) at 45┬Āmg/kg, freshly dissolved in 0.1M sterile sodium citrate, pH 4.5, in 6-week-old rats weighing 180ŌĆō220┬Āg. The rats were considered diabetic if their blood glucose levels were above 200┬Āmg/dL at 48 hours after STZ injection. Control rats were given an equivalent amount of saline via the tail vein. The rats were sacrificed 48 hours, 4 weeks, and 10 weeks after the induction of diabetes. Each group consisted of four animals. Both kidneys were removed and used for the immunofluorescence assessment.
Cell culture of rat glomerular epithelial cells and mouse podocytes
Rat glomerular epithelial cells (GEpCs), cloned from primary rat glomerular cultures by Kreisberg [13], were characterized by a sensitivity to puromycin amino nucleoside, positive staining for Heymann antigen (gp330) and podocalyxin, and negative staining for factor VIII [13], [14], [15]. GEpCs were maintained as previously described [15]. Experiments were performed with cells between passages 15 and 18. Conditionally immortalized mouse podocytes were kindly provided by Dr Peter Mundel (University of Harvard, Boston, MA, USA) and were cultured and differentiated for at least 2 weeks as described previously [16].
Preparation of culture additives and treatment conditions
Cells were serum-deprived to reduce the background 24 hours prior to each experiment and then exposed to glucose and/or AGEs. Rat GEpCs or mouse podocytes were incubated in culture medium containing either 5mM glucose (normal glucose) or 30mM glucose (HG) without insulin. AGEs were produced using the technique previously described by Ha et al [15]. To imitate the long-term diabetic condition, AGEs were added (5┬Ā┬Ąg/mL), and controls were established using unmodified bovine serum albumin (5┬Ā┬Ąg/mL). To exclude the effect of additionally produced glycated proteins during culturing, incubation did not last longer than 48 hours. The fetal bovine serum was reduced to 0.5% at the last medium change to reduce the background caused by any humoral effects of the serum prior to protein and RNA extraction. For identification purposes, AGEs and bovine serum albumin were denoted ŌĆ£AŌĆØ and ŌĆ£BŌĆØ, and glucose at 5mM and 30mM was denoted by ŌĆ£5ŌĆØ and ŌĆ£30ŌĆØ, respectively. The meaning of each condition has been described previously [15]. For AMPK activation, 5-aminoimidazole-4-carboxamide-1╬▓-riboside (AICAR; Merck KGaA, Darmstadt, Germany) and metformin (Daewoong Pharmaceutical Co., Seoul, Korea) treatment was applied using concentrations of 0.5mM and 2mM, respectively, for 24 hours. For AMPK inhibition, compound C (Merck KGaA) was added to the medium at a concentration of 5┬ĄM for 24 hours.
Immunoblotting analysis for AMPK
The confluently grown cell layers were incubated with additives for various durations, extracted in protein extraction solution (PRO-PREP; Intron, Seongnam, Korea) containing phenylmethylsulfonyl fluoride, ethylendiamine tetraacetic acid (EDTA), pepstatin A, leupeptin, and aprotinin, and then the protein concentrations were determined as previously described [16]. For the immunoblotting assay, 25┬Ā┬Ąg of boiled extracts was applied to 10% sodium dodecyl sulfateŌĆōpolyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). Then, the membranes were air-dried and blocked in 3% fat-free milk prior to incubation with monoclonal rabbit antiphospho-AMPK╬▒1Ō¦Ė2 (Thr172) (Cell Signaling Technology, Danvers, MA, USA) and polyclonal rabbit anti-AMPK╬▒1Ō¦Ė2(Santa Cruz Biotechnology, Santa Cruz, CA, USA). As a loading control, anti-╬▓-tubulin antibody (Santa Cruz Biotechnology) was also used. After incubation with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology), bands were detected using the ECL chemiluminescence system (Amersham Biotech Ltd., Bucks, UK). The density values were expressed as % of control (B5).
Immunofluorescence staining
Podocytes that were grown on type I collagen-coated glass coverslips incubated for 24 hours were fixed in 4% paraformaldehyde, permeabilized in phosphate buffer solution, blocked with 10% normal goat serum, and labeled with monoclonal rabbit antiphospho-AMPK╬▒1Ō¦Ė2 (Thr172) (Cell Signaling). F-actin was visualized with TRITC-phalloidin (Sigma Chemical Co.). Primary antibody-bound specimens were incubated with 1:500 (v/v) Alexa 488 for green and Alexa 594 for red (Invitrogen, Eugene, OR, USA) conjugated secondary antibodies at room temperature for 1 hour. Coverslips were mounted in an aqueous mounting medium and viewed with a fluorescence microscope (BX51; Olympus, Tokyo, Japan).The fluorescence intensities were evaluated and calculated using the public domain image-processing program IMAGEJ 1.46r (National Institutes of Health, Bethesda, MD, USA).
Results
Diabetic conditions relocate AMPKs in podocytes
In diabetic renal tissue, we found that phospho-AMPK╬▒ stainings were located along the glomerular capillary loop, the intensities of which were significantly reduced according to the diabetic duration (Fig. 1).
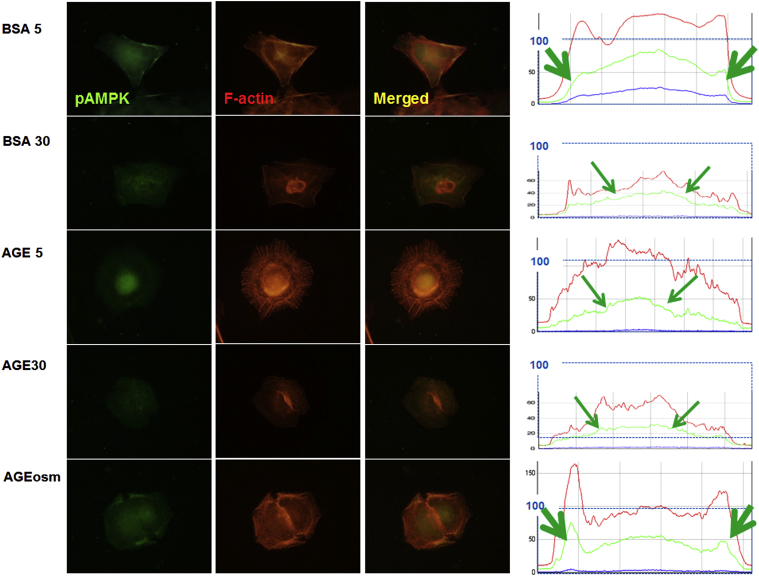
In cultured rat GEpCs, we found that phospho-AMPK╬▒ stainings were diffusely located along with F-actin in the peripheral cytoplasm in the control (B5) and osmotic control (Aosm) (Fig. 2). However, diabetic conditions (B30, A5, and A30) suppressed the intensities of phospho-AMPK╬▒, especially in peripheral cytoplasmic areas, which were somewhat dissociated from the rearranged F-actin fibers (Fig. 2).
Diabetic conditions decrease the phosphorylation of AMPK
HG, including B30 and A30, but not A5, reduced the AMPK╬▒ phosphorylation of rat GEpCs in a time-dependent manner by 36.2% and 25.9%, respectively, after correcting for the total AMPK (n=3) and similarly by 31.5% and 28.1%, respectively, after correcting for the ╬▓-tubulin levels, compared with the control (B5) (n=3, Fig. 3A). The longer their exposure durations were, the more prominent suppressions by AGEs were found.
In mouse podocytes, neither AGEs (A5) nor HG (B30) changed the AMPK╬▒ phosphorylation; however, both AGEs and HG (A30) reduced AMPK╬▒ phosphorylation additively at 24 hours by 45.8% (P<0.01) after correcting for the total AMPK and similarly by 46.1% after correcting for the ╬▓-tubulin levels compared with the control (B5) (n=3, Fig. 3B). There was also a somewhat reduced phospho-AMPK╬▒ response in the osmotic control of AGEs (Aosm), which means that AGEsŌĆörather than HGŌĆöhave a major suppressive effect on mouse podocytes. At 48 hours, A30 reduced the AMPK╬▒ phosphorylation by 42.9% (P<0.05) after correcting for the total AMPK. After correcting for the ╬▓-tubulin levels, AGEs (A5 and A30)ŌĆörather than HGŌĆöalso reduced the AMPK╬▒ phosphorylation by 40.6% (P<0.05) and 54.8% (P<0.01), respectively, compared with the control (B5) (n=3, Fig. 3B). Therefore, diabetic conditions suppressed AMPK╬▒ phosphorylation, although the similar effects were somewhat cell-specific.
AMPK activators ameliorate the changes in phospho-AMPK╬▒ in podocytes
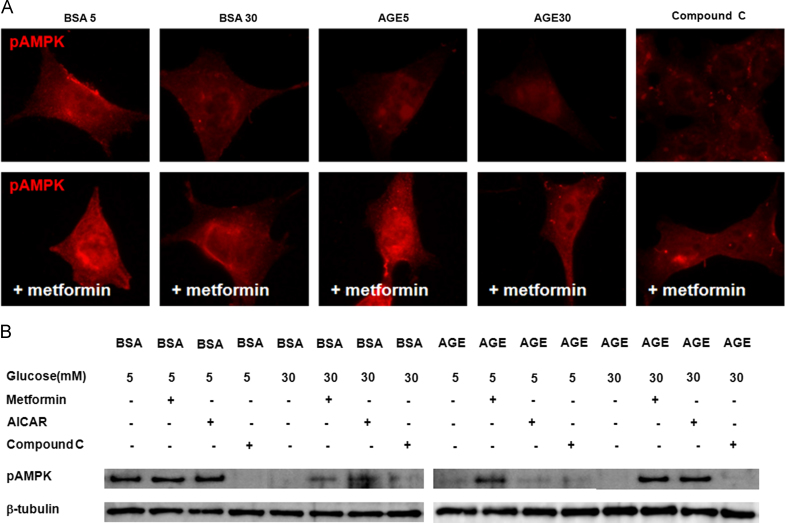
To further assess the involvement of AMPK modulating agents in the regulation of AMPK, we incubated cells with applied AMPK activators, AICAR and metformin, at concentrations of 0.5mM and 2mM, respectively. Similar to the data shown in Fig. 2, diabetic conditions (B30, A5, and A30) and compound C, an AMPK inhibitor, decreased and relocalized the immunofluorescent intensities of phospho-AMPK╬▒ to the internal cytoplasm and to peri- and intranuclear areas of mouse podocytes. Such changes were prevented by an AMPK activator, metformin (Fig. 4A). In the Western blotting assay, diabetic conditions (B30, A5, and A30) and compound C suppressed phospho-AMPK╬▒ in mouse podocytes, similar to that shown in Fig. 3B, a condition that was reversed by AICAR and metformin (Fig. 4B). Therefore, diabetic conditions suppressed and relocalized phospho-AMPK╬▒ in podocytes, and AMPK activators modulated the podocyte injury induced by diabetic conditions.
Discussion
A number of physiological processes have been shown to activate AMPK, including conditions that lead to an increase in the intracellular AMP/ATP ratio (such as heat shock, hypoxia/anoxia, ischemia, and glucose deprivation) and the calcium concentration, along with the action of various hormones, cytokines, and adipokines [4], [5], [6]. The activated form of AMPK is responsible for metabolic changes via the phosphorylation of various downstream substrates. The net effect is a change in local and whole-body energy utilization from an energy-consuming state to an energy-producing state to restore the energy balance. These findings, coupled with reports that AMPK in the muscle is activated in response to exercise [17], [18], have led to an intense interest in developing AMPK activators as potential therapies for type 2 diabetes and obesity [19].
AMPK and its activator, adiponectin receptor ADIPOR1, localized at the plasma membranes of the glomerular cells, including endothelium, mesangial cells, and podocytes, as well as on parietal epithelial cells. Either adiponectin or AICAR led to the activation of catalytic AMPK by phosphorylation in isolated rat glomeruli [20]. This suggests that the activation of glomerular AMPK by adiponectin or AICAR might play an important role in the control of oxidative stress and cell survival within the glomerulus.
It has been reported that AMPK activity is reduced in the diabetic kidney [21], [22], [23], [24], [25]; however, the reduced AMPK activity in the diabetic kidney does not appear to be related to altered AMP or ATP levels [21]. Guo et al [21] have correlated the reduced AMPK activity in STZ-induced diabetic rats to reduced adiponectin levels, increased expression of connective tissue growth factor, and increased triglyceride accumulation. The changes in AMPK activity in the diabetic kidney do not appear to be related to altered AMP or ATP levels. Cammisotto et al [22] have demonstrated that the expression levels of ADIPOR1, AMPK╬▒(1), AMPK╬▒(2), and AMPK╬▓(2) were increased in STZ-treated diabetic rats, whereas phosphorylated active AMPK levels were strongly decreased. They also demonstrated that adiponectin through luminal ADIPOR1 activates AMPK, leading to the inhibition of glycogen synthase.
Regarding podocytes, Lee et al [23] have shown that reduced AMPK╬▒ subunit threonine (Thr) 172 phosphorylation in the rat diabetic kidney and HG-treated cultured rat GEpCs, the same cells as ours, was associated with increased activation of hypertrophy marker proteins, including 4E binding protein 1 and eukaryotic elongation factor 2, and mammalian target of rapamycin (mTOR). Metformin and AICAR increased renal AMPK phosphorylation, reversed mTOR activation, and inhibited renal hypertrophy, without affecting hyperglycemia in these models. We found the suppression of phospho-AMPK╬▒ not only by HG but also by AGE in this experiment. In diabetic models, Eid et al [24] have also found that high glucose diabetic conditions in vitro and in vivo inactivated AMPK, upregulated Nox4, enhanced NAD(P)H oxidase activity, and subsequently induced podocyte apoptosis, which were inhibited by AMPK activation. In the following experiment, they found that HG also activated mTOR, which was downstream of AMPK [25]. Recently, we found that angiotensin II, an important inducer of diabetic injury, suppressed AMPK╬▒ phosphorylation in podocytes via angiotensin II type 1 receptor and MAPK signaling, which may cause podocyte dysfunction [26]. Piwkowska et al [27] have also shown that metformin decreased the production of reactive oxygen species, another major diabetic injury mediator, through a reduction of NAD(P)H oxidase activity and increased AMPK activity. Therefore, AMPK activator could prevent glucose-induced oxidative stress in podocytes.
Reduced AMPK activity in the diabetic kidney has also been linked to increased triglyceride accumulation because of the reduced inhibitory phosphorylation of acetyl-CoA carboxylase [21]. The increased triglyceride accumulation in the diabetic kidney appears to increase the expression of connective tissue growth factor, which is another mechanism that contributes to diabetes-induced renal hypertrophy [21]. In addition, as described above, reduced AMPK activity in the diabetic kidney has been associated within creased renal tubular glycogen accumulation [22]. In an obesity animal model, Sharma et al [28] have also found that a low serum level of adiponectin is associated with albuminuria and that adiponectin treatment in cultured podocytes increased the activity of AMPK and reduced podocyte permeability to albumin and podocyte dysfunction. Furthermore, adiponectin knockout mice exhibit increased albuminuria and fusion of podocyte foot processes [28]. They assumed that such effects appeared to be caused by a reduction of oxidative stress, as adiponectin and AMPK activation both reduced the protein levels of NAD(P)H oxidase 4 (Nox4) in podocytes [28].
Although the mechanism for the reduced AMPK activity in the diabetic nephropathy is unclear, adiponectin may be associated with AMPK activation, and reduced AMPK activity by mTOR signaling, angiotensin II, and oxidative stress in diabetic conditions would induce renal hypertrophy and apoptosis. A similar suppression of AMPK activity was observed in podocytes; this would be an important mechanism and a therapeutic target for treating podocyte injuries induced by diabetic conditions.
Another notable finding of our study is that diabetic conditions induce the relocation of phospho-AMPK╬▒ from the peripheral cytoplasm to the internal cytoplasm and peri- and intranuclear areas in podocytes. Although we could not find any results on the relocation of podocytes AMPK╬▒ in pathologic conditions, we suggest that its relocation might be caused by the defect in trafficking; therefore, it could induce the decrease in its action at a signaling area in addition to the reduced AMPK activity. Further observation on the cellular fractional changes of AMPK╬▒ is necessary to elucidate the meaning of its relocalization.
In conclusion, we demonstrated that diabetic conditions suppressed and relocalized phospho-AMPK╬▒ in podocytes, and AMPK activators modulated the podocyte injury induced by diabetic conditions. Therefore, the activation of AMPK signaling might serve as a new therapeutic target for the treatment of diabetic podocytopathy.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









