



Introduction
Apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1) is a ubiquitously expressed multipotent protein that plays an essential role in cellular response to oxidative stress. APE1/Ref-1 (also known as HAP1 or APEX) is a protein composed of 318 amino acids including a redox region and a DNA repair region. It is an essential component in maintaining genome stability, serving as an essential master regulator of the stress response [1]. APE1/Ref-1 is a dual-function protein involved both in the base excision repair pathways of DNA lesions, acting as the major apurinic/apyrimidinic endonuclease, as well as in the eukaryotic transcriptional regulation of gene expression [2,3]. APE1/Ref-1 functions to activate many transcription factors including p53, nuclear factor kappa B (NF-κB), hypoxia-inducible factor (HIF) 1-α, and paired box 5 [2,4]. Moreover, APE1/Ref-1 may be important in the cellular response to oxidative changes triggered during apoptosis. APE1/Ref-1 is localized predominantly in the nucleus and has also been detected in the mitochondria and cytoplasm [5]. The movement of APE1/Ref-1 from the nucleus to the cytoplasm is mediated by an N-terminal nuclear export signal and a C-terminal nuclear localization signal (NLS) [6,7]. Importantly, this subcellular localization is dynamically regulated. APE1/Ref-1 rapidly translocates from the nucleus to the cytoplasm in response to reactive oxygen species (ROS) [8–10]. These two biological activities are located in two functionally distinct domains. The N-terminus, containing the NLS region, is principally devoted to redox activity through Cys65, while the C-terminus exerts enzymatic activity towards the basic sites of DNA [11].
Accumulating evidence has demonstrated that the heterogeneity of APE1/Ref-1 expression patterns is linked to different pathological conditions ranging from metabolic to differentiative disorders, including cancer and neurodegenerative diseases. Different kinds of human tumors are characterized by alterations in the subcellular distribution of APE1/Ref-1 with respect non-tumoral tissue [4,12]. The expression status of APE1/Ref-1 is altered in numerous cancers including prostate, lung, colon, and ovarian tumors. In addition, alterations in APE1/Ref-1 expression and mutations in the APE1/Ref-1 gene have been detected in patients with a variety of neurodegenerative diseases [2,13–18]. However, the pathophysiological roles of APE1/Ref-1 in kidney disease remain unclear.
In the present study, we evaluated whether APE1/Ref-1 was involved in ischemia-reperfusion (I/R)–induced kidney injury and whether APE1/Ref-1 played roles in H2O2-treated human proximal epithelial tubule (HK-2) cells. We also investigated whether APE1/Ref-1 overexpression exhibited cytoprotective or cytotoxic effects against H2O2-mediated apoptosis.
Methods
Animal experiments
The animal experiments were approved by the Animal Care Regulations Committee of the Chonnam National University Medical School (No. CNUH IACUC-18010), and the protocols conformed to the institutional guidelines for experimental animal care and use. Eight-week-old male C57BL6 mice were purchased from Samtako. Kidney I/R injuries were established by clamping both renal arteries for 20 minutes followed by 2 days of reperfusion. During the period of ischemia, body temperature was maintained by placing the rats on a 37 °C heating pad. Following removal of the clamps, the kidneys were inspected for 1 minute for the restoration of blood flow, as noted by a return to their original color, prior to closure of the abdomen. Sham-operated mice received identical surgical procedures with the exception of the microaneurysm clamps. The mice were sacrificed 2 days after reperfusion, blood was collected, and kidney tissue was divided for subsequent protein extraction.
Cell culture and reagents
HK-2 cells were passaged every 3–4 days in 100-mm dishes containing Dulbecco’s modified Eagle’s medium/F-12 medium supplemented with 10% fetal bovine serum (FBS), 100-U/mL penicillin, and 100-μg/mL streptomycin (Sigma). When the cells reached 70% to 80% confluence, they were detached with TrypLE Express (Invitrogen), centrifuged at 250 ×g for 3 minutes, replated, and maintained at 37 °C in an incubator containing 5% CO2 atmosphere. For experimental use, the HK-2 cells were plated in 60-mm dishes in medium containing 10% FBS for 24 hours and then treated with H2O2.
Histologic analysis
Kidney tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 4 μm-thick sections. To assess histological morphology, immunohistochemical staining was performed using indicated antibodies and horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G secondary antibodies (Dako). The stained sections were imaged with a Nikon Eclipse Ni-U microscope. The quantitative analysis of the stained sections was performed using imageJ software (National Institutes of Health).
Confocal laser microscopy
Left kidneys of I/R and control mice were collected for immunofluorescence analyses. Tissue samples were prepared through deparaffination and hydration, and the cells fixed in 4% paraformaldehyde were prepared and blocked at room temperature for 2 hours. Rabbit or mouse monoclonal antibodies against Lotus tetragonolobus lectin (Vector Laboratories), calbindin-D28K (Invitrogen), Tamm-Horsfall protein (AbD serotec), and aquaporin-2 (Santa Cruz Biotechnology) were diluted 1:100 in blocking buffer and applied at 4 °C for 24 hours. After washing, the secondary antibody was diluted 1:200 in blocking buffer and applied at room temperature for 2 hours. After washing, coverslips were mounted onto microslides using a ProLong Gold Antifate Reagent with DAPI (Life Technologies).
Protein extraction
The kidneys were homogenized in ice-cold isolation solution containing 0.3-M sucrose, 25-mM imidazole, 1-mM ethylenediaminetetraacetic acid (EDTA), 8.5-μM leupeptin, and 1-mM phenylmethylsulfonyl fluoride (PMSF; pH 7.2). The homogenates were centrifuged and the total protein concentration was measured using a Pierce BCA protein assay kit (Thermo Fisher Scientific). All the samples were adjusted with isolation solution to normalize protein concentration and stored at –20 °C. HK-2 cells were harvested, washed twice with ice-cold phosphate-buffered saline (PBS), resuspended in lysis buffer (20-mM Tris-HCl [pH 7.4], 0.01-mM EDTA, 150-mM NaCl, 1-mM PMSF, 1-μg/mL leupeptin, and 1-mM Na3VO4), and briefly sonicated. After centrifugation, the supernatants were prepared as protein extracts and the protein concentrations were measured (Pierce BCA protein assay reagent kit).
Western blot analysis
Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 9% or 12% gels. The proteins were electrophoretically transferred onto nitrocellulose membranes using a Bio-Rad Mini Protean II apparatus (Bio-Rad). The blots were blocked with 5% milk in PBS-T (80-mM Na2HPO4, 20-mM NaH2PO4, 100-mM NaCl, and 0.1% Tween-20 [pH 7.5]) for 2 hours. Anti-Bcl-2 (3498), anti-Bax (2772), anti-cleaved caspase 3 (9661), anti-phospho extracellular signal-regulated kinase (p-ERK) (9101), anti-phospho-p38 mitogen-activated protein kinase (p-p38 MAPK) (4631), and anti-phospho JNK (p-JNK) (9251) antibodies (Cell Signaling Technology) were used. Phospho-NF-κB p65 (sc-33020; Santa Cruz Biotechnology), anti-NF-κB p65 (8242; Cell Signaling Technology), anti-IκBα (SC-1643; Santa Cruz Biotechnology), and anti-β-actin (a5316; Sigma) antibodies were diluted in blocking buffer and incubated with the blots overnight at 4 °C. The blots were then washed and incubated with peroxidase-conjugated secondary antibody (1:3,000). The selected bands were scanned (GS-700 Imaging Densitometry; Bio-Rad) and the density was determined (Molecular Analyst version 1.5; Bio-Rad).
Stable cell lines
To generate APE1/Ref-1 overexpressing cell lines, HK-2 cells were transfected with 2 μg of empty vector (Mock) or APE1/Ref-1 DNA using 6 μL of FuGENE HD reagent (Promega) in antibiotic-free DMEM-F12. Starting 1 day after transfection, transfectants were selected in DMEM-F12 containing 800-μg/mL zeocin which was refreshed every 3 days for 2 weeks. Colonies surviving in the selection medium were collected and sequentially plated in 48-, 12-, and 6-well plates and then in 60- and 100-mm dishes. Total cellular extracts were analyzed for APE1/Ref-1 expression by immunoblotting, thus revealing the expression of the ectopic flagged forms of wild type and the mutant form of the protein.
Small interfering RNA knockdown of APE1/Ref-1
RNA interference of APE1/Ref-1 was performed using an APE1/Ref-1–specific small interfering RNA (siRNA) from Ambion’s siRNA Target Finder Program: Ape1/Ref-1 siRNA (534 bp from the ATG), 5-GUCUGGUACGACUGGAGUAtt-3 (sense), and 5-UACUCC AGUCGUACCAGACtt-3 (antisense). siRNAs were prepared using a transcription-based method with a Silencer siRNA construction kit (Ambion).
Apoptosis assay
The number of apoptotic cells was quantified using the Ezway Annexin V-FITC apoptosis detection kit (KOMA Biotech) according to the manufacturer’s protocol. Cells were stained with Annexin V-FITC dye. Fluorescent intensity was measured by FACSCalibur flow cytometry (BD Biosciences).
Promotor activity of NF-κB
The transcriptional regulation of NF-κB was examined by the transient transfection of an NF-κB promoter-luciferase reporter construct (pGL3-NF-κB). HK-2 cells (5 × 105) were seeded and grown until they reached 60% to 70% confluence, and pGL3-NF-κB wild-type and pGL3-empty were transfected into the cells using FuGene HD reagent according to the manufacturer’s protocol. The pRL-null plasmid encoding Renilla luciferase was included in all the samples to monitor transfection efficiency. At 24 hours after transfection, the levels of Firefly and Renilla luciferase activity were measured sequentially from a single sample using the Dual-Glo luciferase assay system (Promega). Firefly luciferase activity was normalized to Renilla activity and the relative amount of luciferase activity in the untreated cells.
Electrophoretic mobility shift assay
Nuclear extracts of the HK-2 cells were prepared with the NE-PER nuclear extraction reagent (Pierce Biotechnology). The biotin-labeled NF-κB oligonucleotide sequence was 5′-biotin-AGTTGAGGGGACTTTCCCAGGC-3′. The binding reactions contained 10 µg of the nuclear extract protein, buffer (10-mM Tris [pH 7.5], 50-mM KCl, 5-mM MgCl2, 1-mM dithiothreitol, 0.05% Nonidet P-40, and 2.5% glycerol), 1 µg of poly (dI-dC), and 2-nM biotin-labeled DNA. The reactions were incubated at 23 °C for 20 minutes. The competition reactions were performed by adding 10-fold excess unlabeled double-stranded NF-κB consensus oligonucleotides to the reaction mixture. The reactions were electrophoresed on a 6% precasted Tris-borate–EDTA gel (Invitrogen) at 100 V for 1 hour 30 minutes in a 100-mM Tris-borate–EDTA buffer. The reactions were then transferred to a nylon membrane. The biotin-labeled DNA was detected with a LightShift chemiluminescent electrophoretic mobility shift assay kit (Pierce Biotechnology).
Statistical analysis
The results are expressed as the mean ± standard error of mean. Multiple comparisons among the three groups were performed using one-way analysis of variance and post hoc Tukey honestly significant difference tests. Differences with the p-values of <0.05 were considered significant.
Results
APE1/Ref-1 expression in in vivo ischemia-reperfusion–induced mouse kidney injuries
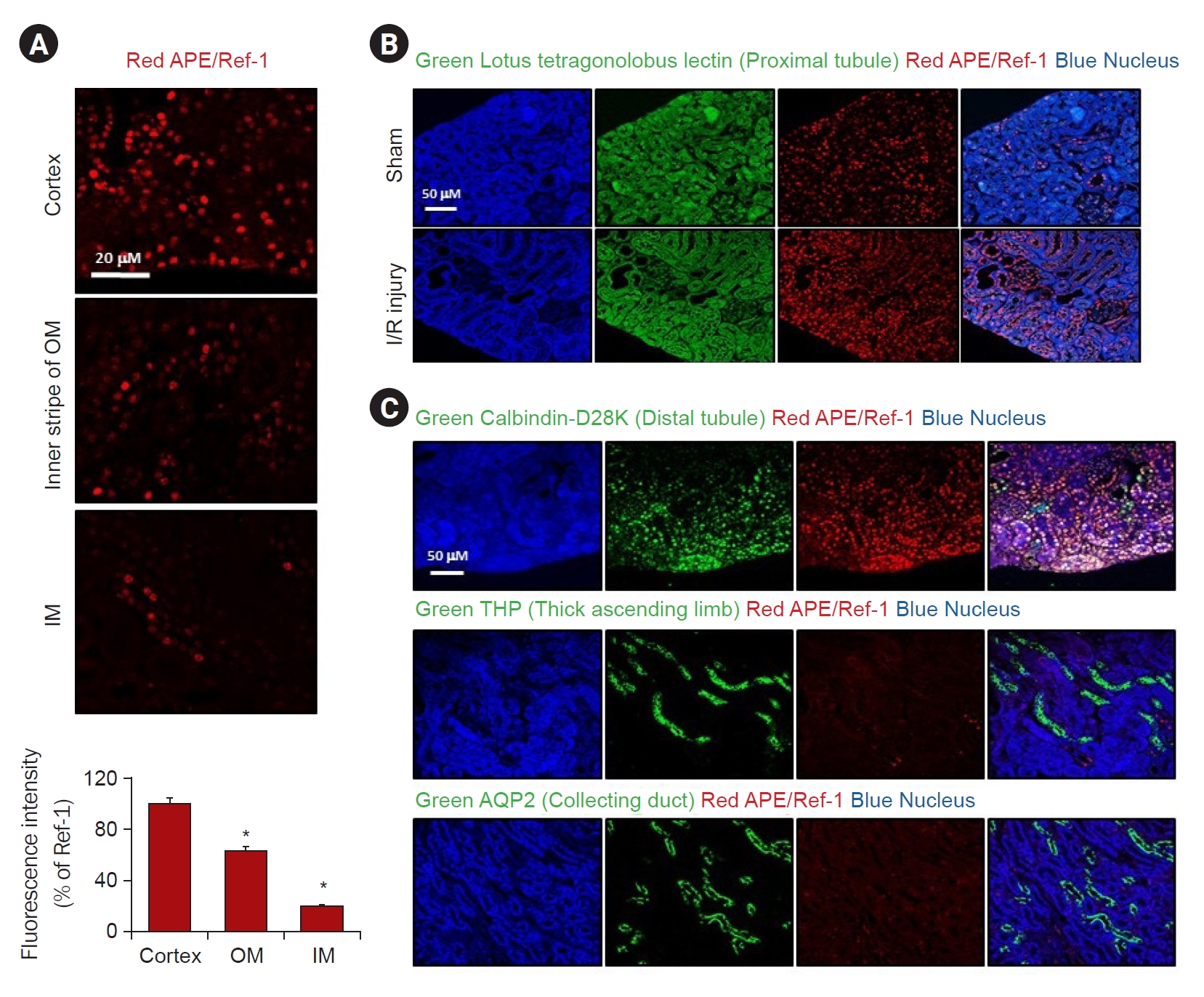
Fig. 1 shows the expression pattern of APE1/Ref-1 according to mouse I/R injury. The fluorescence of APE1/Ref-1 expression was highest in the cortex, modest in the outer medulla, and lowest in the inner medulla (Fig. 1A). The expression of APE1/Ref-1 that significantly increased after I/R injury compared to the control was observed by immunofluorescence (Fig. 1B). Fig. 1C shows the observation of the expression of APE1/Ref-1 in each part of the kidney by immunofluorescence. APE1/Ref-1 exhibited high expression in the proximal tubule, but weak expression was observed in the thick ascending limb and collecting duct in the mouse kidney (Fig. 1C).
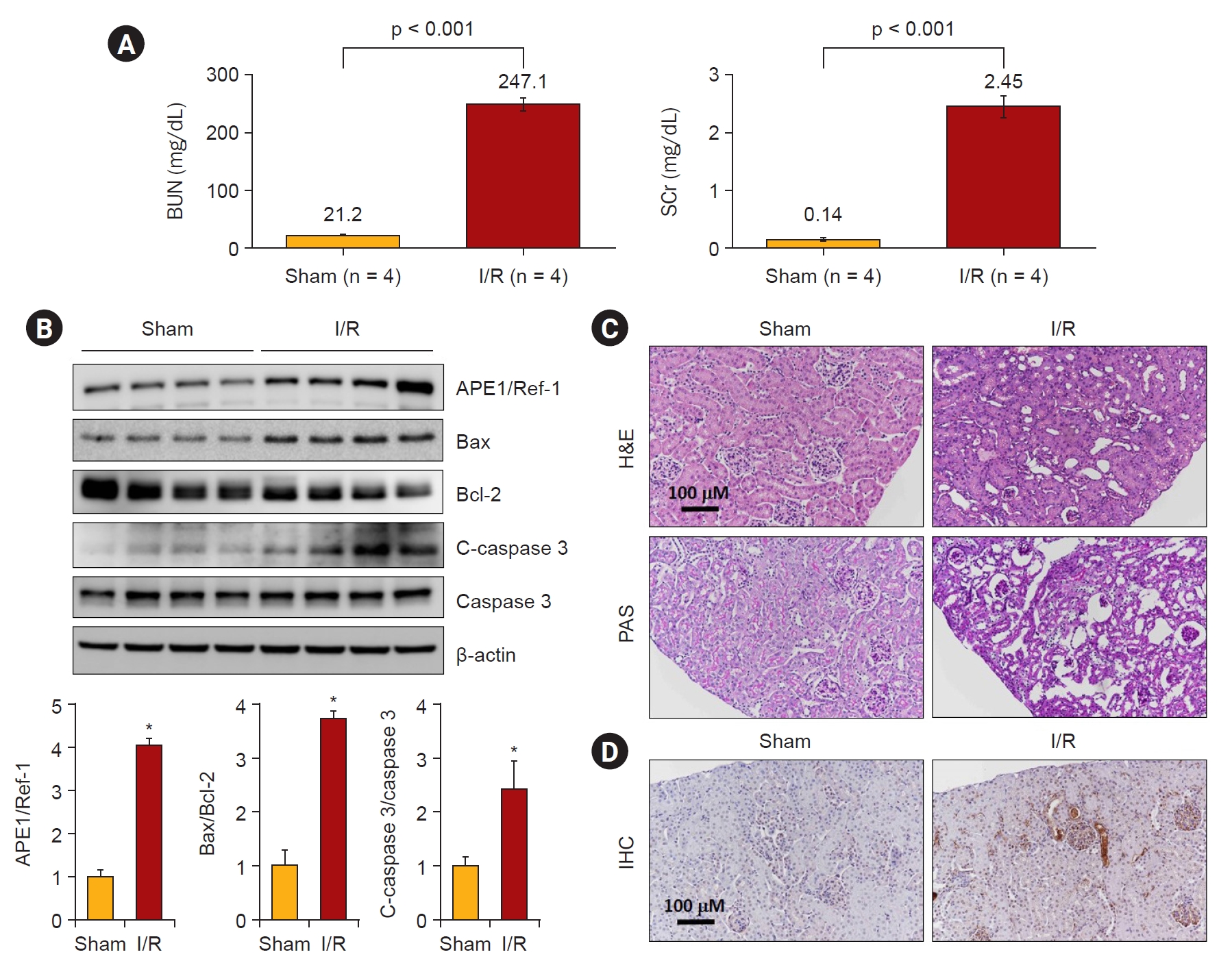
Serum blood urea nitrogen and creatinine levels were significantly increased in I/R injured mice compared to the sham-operated controls (Fig. 2A). Consistent with this finding, the protein expression of APE1/Ref-1 increased after I/R injury (Fig. 2B). In addition, the protein expression levels of Bax and cleaved caspase 3 were increased, whereas Bcl-2 expression was decreased. We observed kidney tissue damage caused by I/R through H&E and PAS staining. There was a markedly increased kidney tubule-interstitial injury tissue in the I/R model compared to the control (Fig. 2C). Immunohistochemistry was performed to determine whether I/R-induced renal tissue damage was associated with the expression of APE1/Ref-1. Compared with the control, it was observed that the expression of APE1/Ref-1 increased in the I/R injury model (Fig. 2D). These results demonstrate that APE1/Ref-1 expression and apoptosis are induced by I/R kidney tissue damage.
Aggravation of H2O2-mediated injury by APE1/Ref-1 overexpression in HK-2 cells in vitro
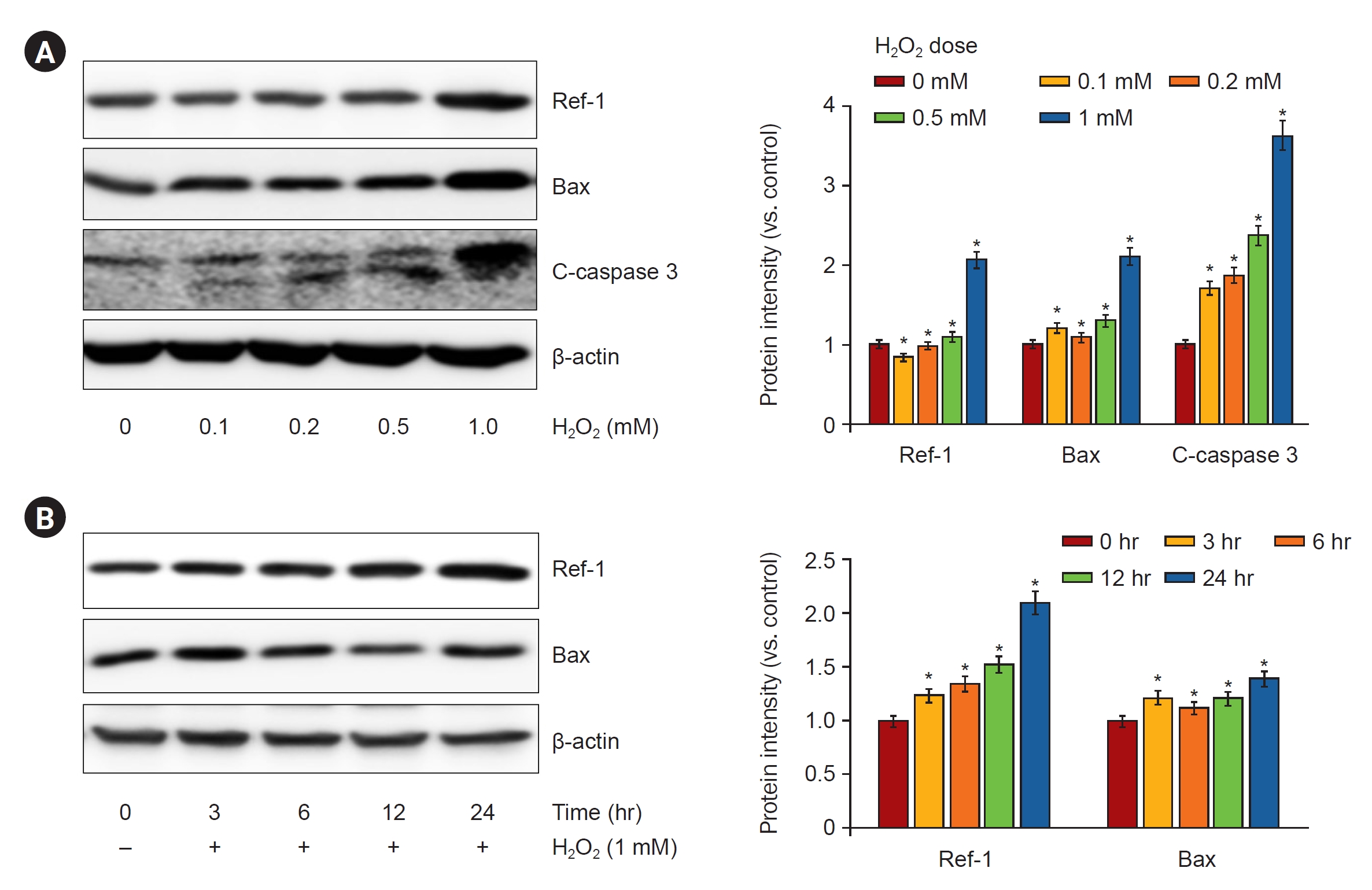
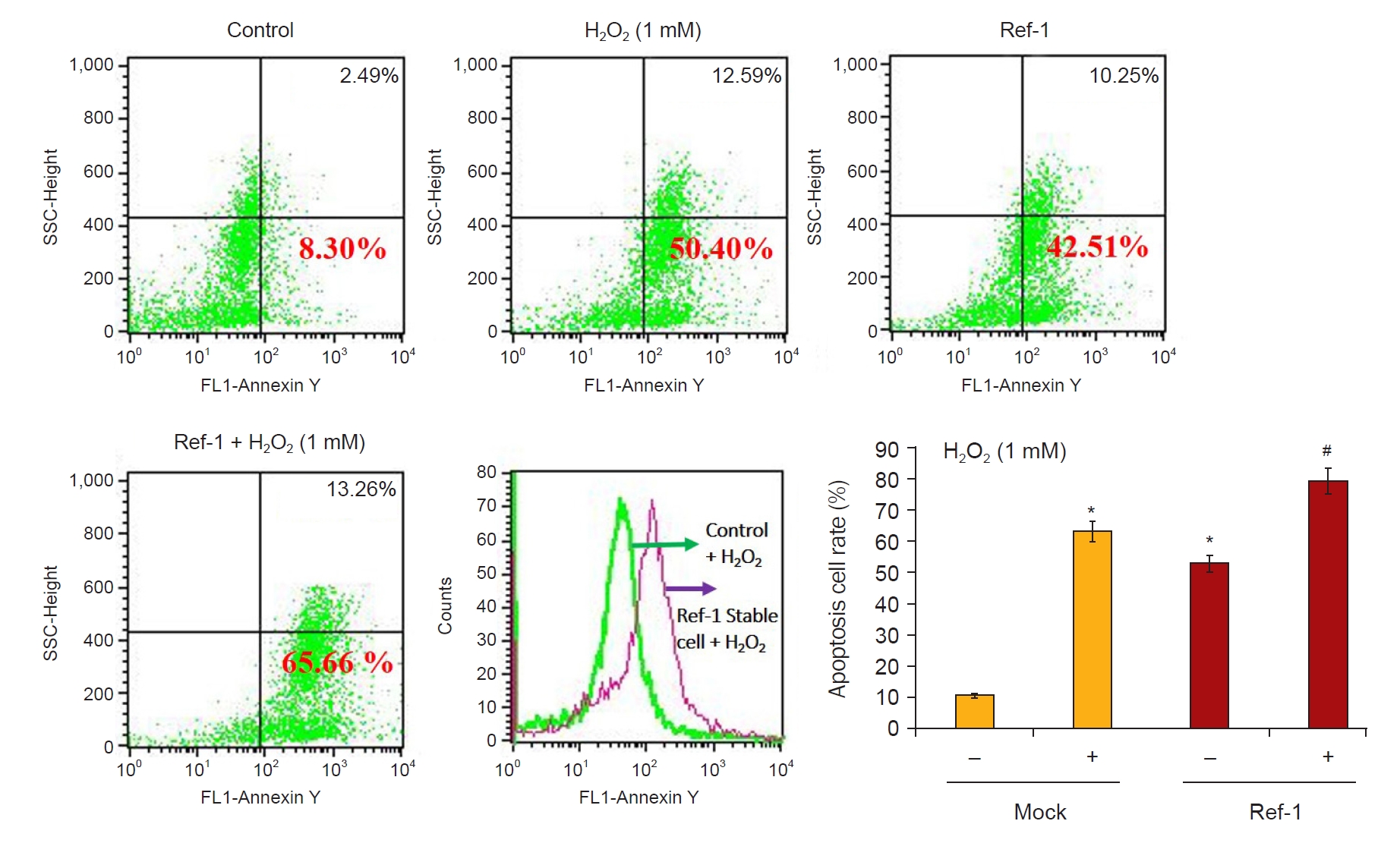
Next, we investigated whether H2O2 treatment increased the expression of APE1/Ref-1 and apoptosis proteins in kidney proximal tubular HK-2 cells. Fig. 3A shows that 0-, 0.1-, 0.2-, 0.5-, and 1-mM H2O2 were treated in a concentration-dependent manner and harvested after 24 hours. APE1/Ref-1, Bax, and cleaved caspase-3 increased according to the concentration. Fig. 3B shows the time-dependent observations of 0, 3, 6, 12, and 24 hours by treatment with 1-mM H2O2. The expression of APE1/Ref-1 and Bax was significantly increased after 24 hours. Next, we evaluated the effects of the overexpression of APE1/Ref-1 in H2O2-treated HK-2 cells in vitro. To determine the physiological effects of APE1/Ref-1, HK-2 cells were stably transfected with an empty vector (Mock) or a plasmid encoding human APE1/Ref-1. Stable cells were selected through the confirmation of the expression of zeocin, which was present in the backbone plasmid pCDNA4. The evaluation of Mock and APE1/Ref-1 stable cells following H2O2-mediated injury revealed decreased cell viability and increased the number of Annexin-V-positive cells in cells stably expressing APE1/Ref-1. Apoptotic cells were defined as Annexin V-FITC-positive cells. This result suggested that APE1/Ref-1 overexpression aggravated H2O2-mediated apoptotic and necrotic cell death (65.7% apoptosis in H2O2-treated HK-2 cells with APE1/Ref-1 overexpression versus 50.4% apoptosis in H2O2-treated HK-2 cells without APE1/Ref-1 overexpression) (Fig. 4).
Proapoptotic effects of APE1/Ref-1
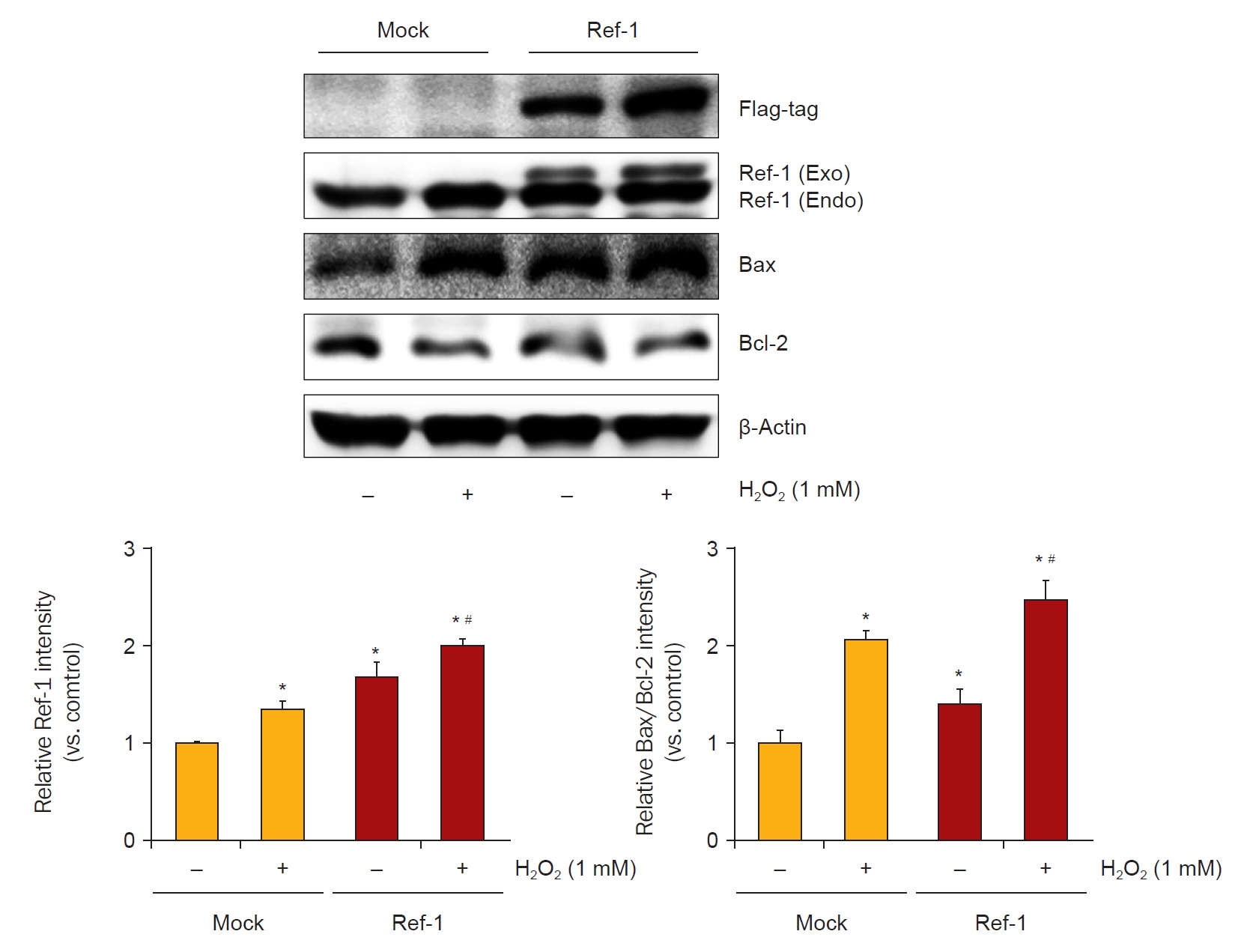
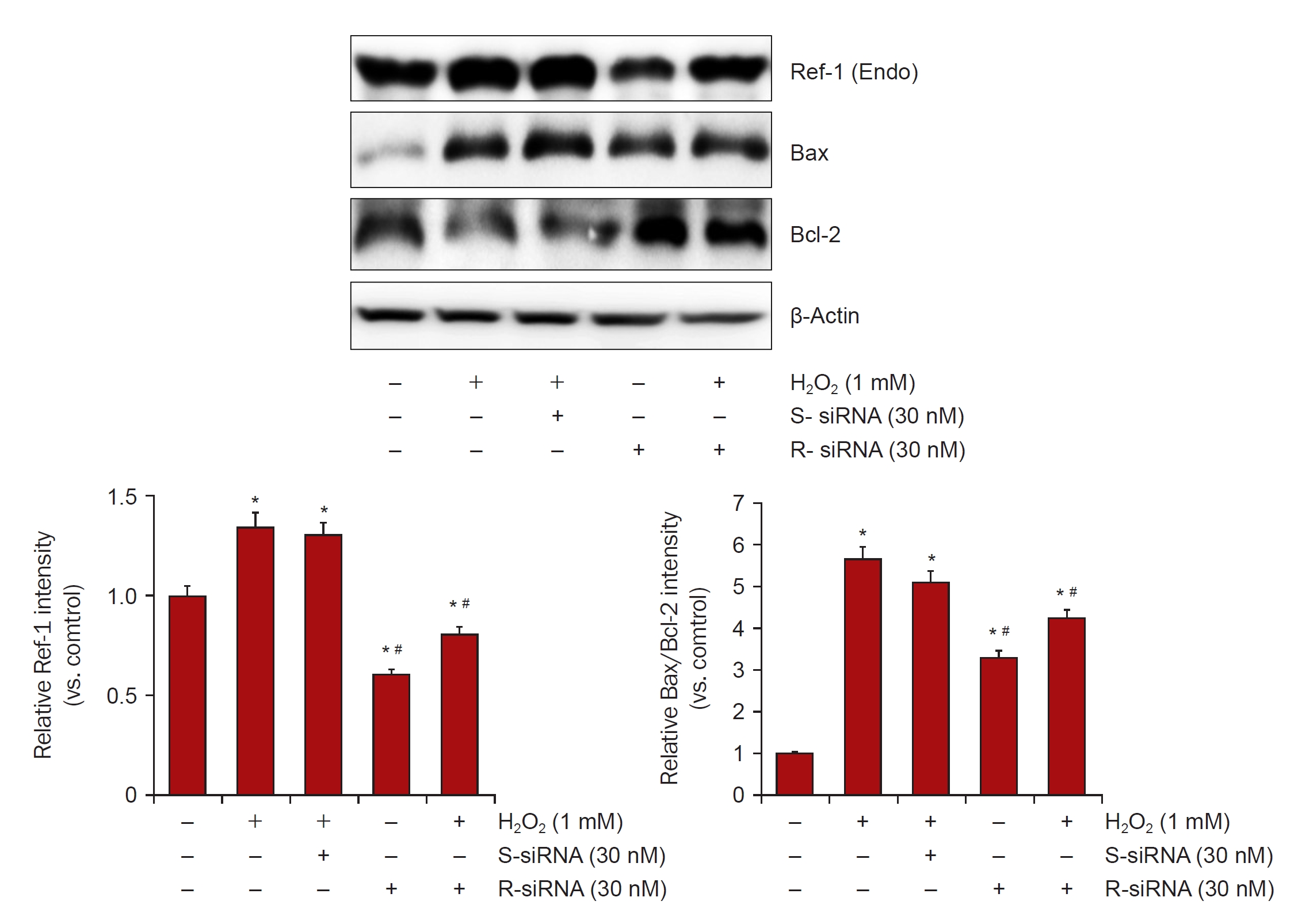
To verify the apoptotic effects of APE1/Ref-1 in injured proximal tubule cells, the expression of proapoptotic proteins was evaluated in H2O2-treated Mock cells and cells stably expressing APE1/Ref-1. The expression level of the Bax/Bcl-2 ratio was increased in APE1/Ref-1 overexpressing cells (Fig. 5), and this effect was abolished by APE1/Ref-1 specific siRNA knockdown (Fig. 6).
Role of APE1/Ref-1 in mitogen-activated protein kinase signaling
Following treatment with 1-mM H2O2, the MAPK pathway components (e.g., extracellular signal-regulated kinase [ERK], c-Jun N-terminal kinase [JNK], and p38) were upregulated in APE1/Ref-1-overexpressing cells compared with Mock cells (Fig. 7A). The phosphorylation of p38, which is involved in the stabilization and mitochondrial accumulation of p38, was higher in H2O2-treated APE1/Ref-1 cells compared to Mock cells. Furthermore, the levels of activated p-ERK1/2 and phospho-JNK1/2 were enhanced in H2O2-treated APE1/Ref-1–overexpressing cells, and these effects were attenuated by transfection with siRNA targeting APE1/Ref-1 (Fig. 7B). Therefore, the involvement of the ERK/p38/JNK axis in apoptosis was associated with APE1/Ref-1 overexpression.
Effects of APE1/Ref-1 on NF-κB signaling
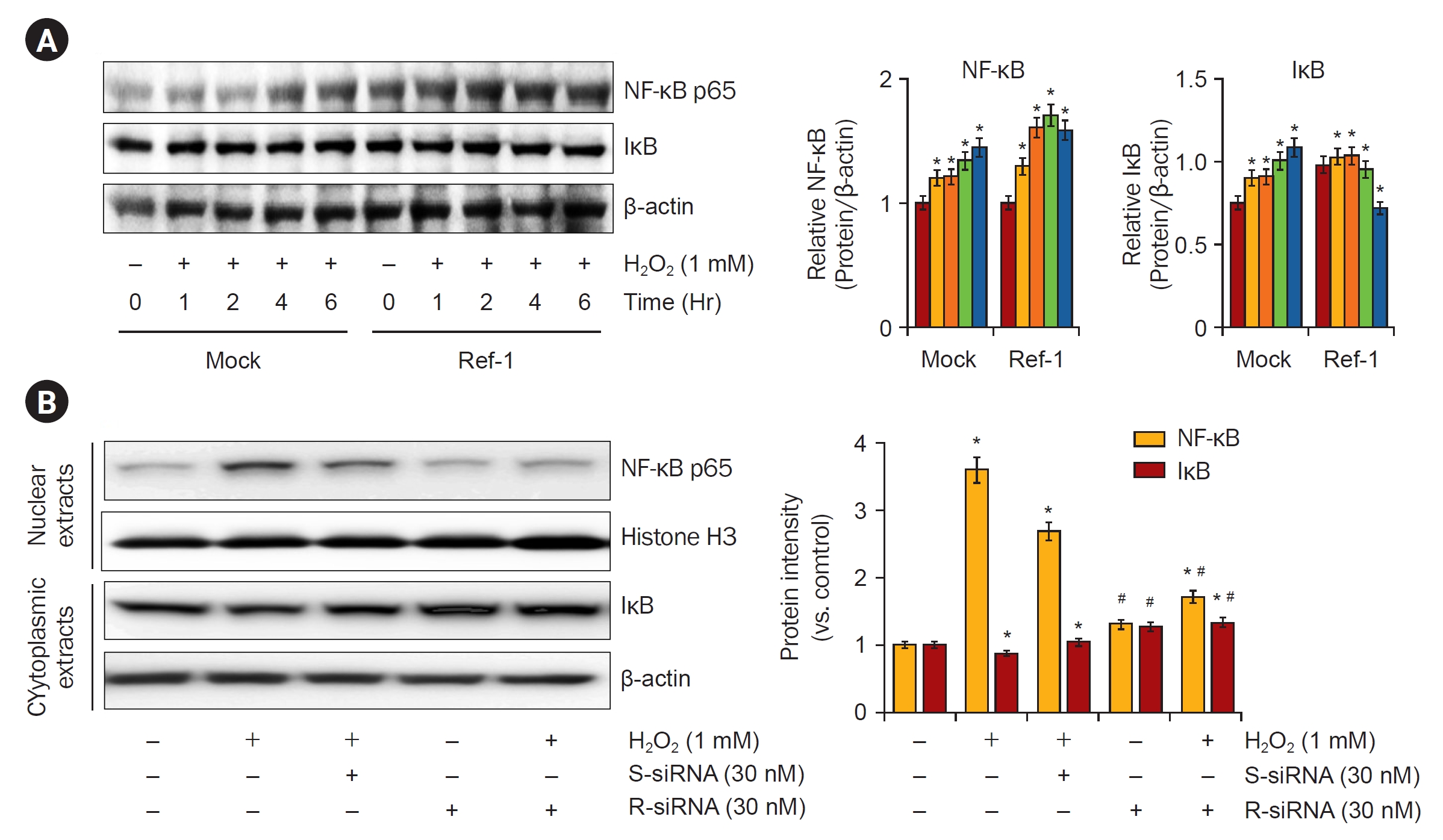
To further analyze the mechanisms through which APE1/Ref-1 was associated with the enhancement of apoptosis, we studied the expression of NF-κB, which is involved in the coordinated induction of genes that encode many stress-responsive and cytotoxic enzymes and related proteins. After treatment with 1-mM H2O2, NF-κB protein expression was observed in a 0, 1-, 2-, 4-, and 6-hour dependent manner. The expression of NF-κB protein was significantly increased in APE1/Ref-1 overexpressing cells compared with Mock cells (Fig. 8A). Next, NF-κB expression by APE1/Ref-1 specific siRNA knockdown was observed. Cells were treated with 30 nM of scrambled siRNA and APE1/Ref-1 siRNA for 24 hours, and then treated with 1-mM H2O2 for 6 hours to obtain nuclear and cytoplasmic proteins, respectively, and NF-κB expression was observed. We observed the decreased expression of NF-κB nucleoproteins transfected with siRNA targeting APE1/Ref-1 (Fig. 8B). These results suggest that the expression of APE/Ref-1 and NF-κB is related to apoptosis.
Effects of APE1/Ref-1 on the transcriptional activation of NF-κB
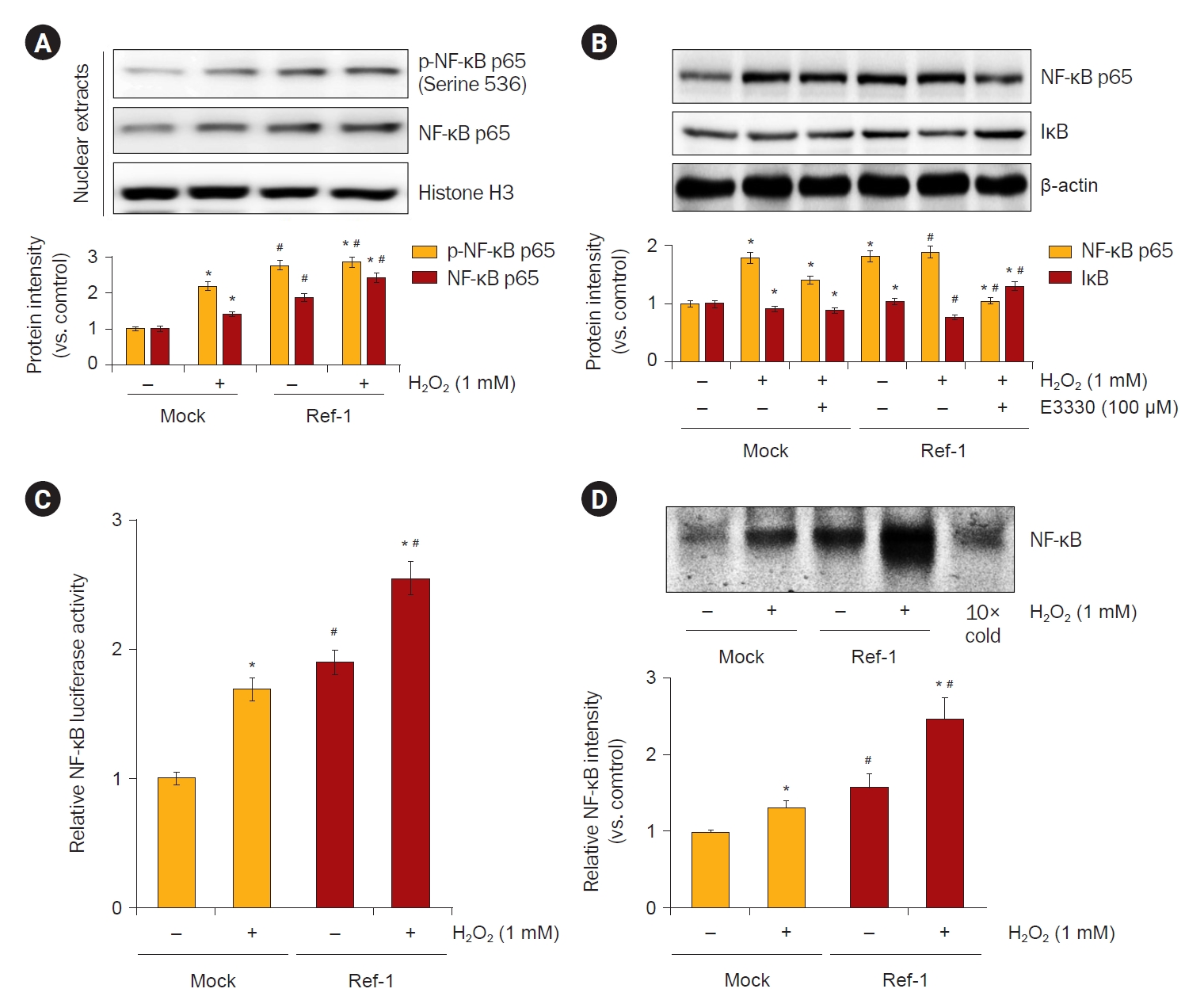
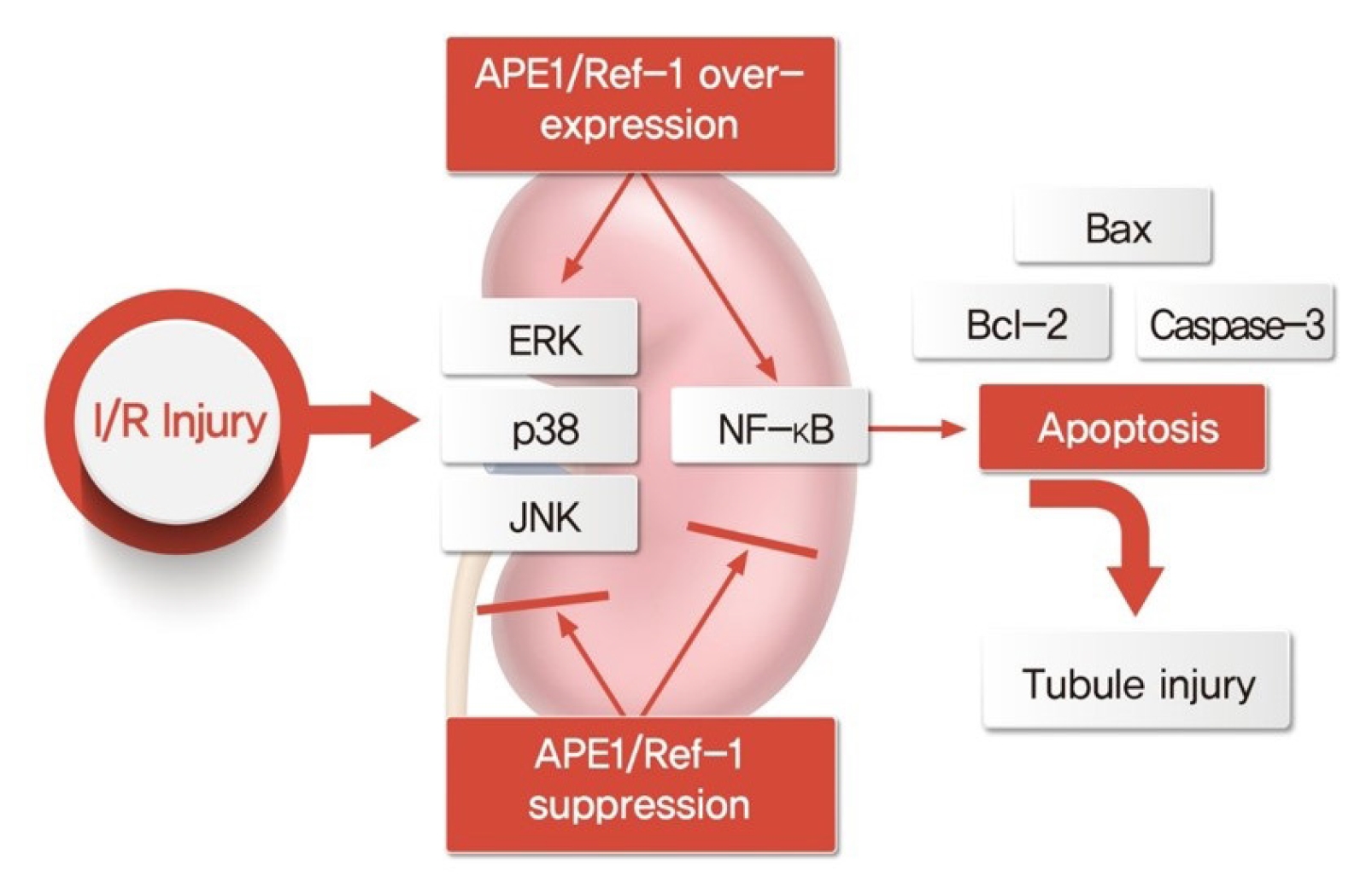
NF-κB is an important transcription factor activating the expression of cyclooxygenase-2 and inducible nitric oxide synthase. Moreover, NF-κB is known to be activated by ROS. Therefore, we examined the role of APE1/Ref-1 in the H2O2-induced transcriptional activation of NF-κB. First, it was observed whether NF-κB phosphorylation increased in nucleoproteins after treatment with 1-mM H2O2. The expression of NF-κB proteins was observed to significantly increase the phosphorylation of NF-κB in APE1/Ref-1 overexpressing cells compared to Mock cells (Fig. 9A). NF-κB is activated by a Cys-65 redox activation reaction that regulates several transcription factors present in the N-terminal region of APE1/Ref-1. We treated cells with an inhibitor of APE1/Ref-1 (E3330) and observed whether NF-κB activity was inhibited. NF-κB was significantly reduced by treatment with 100 μM E3330 (Fig. 9B). The promoter activity of NF-κB was increased following H2O2 exposure in HK-2 cells, and this increase was enhanced by APE1/Ref-1 transfection (Fig. 9C). Nuclear extracts from cells analyzed by electrophoretic mobility shift assays for activated NF-κB confirmed these findings (Fig. 9D). These results suggest that APE1/Ref-1 increased NF-κB pathway activity directly by activating the transcription factor NF-κB. Based on these results, a schematic diagram showing that APE1/Ref-1 induces apoptosis in association with MAPK and NF-κB signaling is presented in Fig. 10.
Discussion
Ischemic stress contributed to the upregulation of APE1/Ref-1 expression in the kidney. In HK-2 cells, oxidative stress induced increased APE1/Ref-1 expression, and APE1/Ref-1 aggravated H2O2-mediated apoptosis in APE1/Ref-1-overexpressing HK-2 cells. Moreover, the Bax/Bcl-2 ratio was increased in APE1/Ref-1-overexpressing cells but was decreased in HK-2 cells transfected with siRNA targeting APE1/Ref-1. We also demonstrated that APE1/Ref-1 was associated with the MAPK pathway and NF-κB signaling. These findings suggest that the upregulation of APE1/Ref-1 is related to apoptosis after oxidative stress and that the inhibition of APE1/Ref-1 could have therapeutic potential in the management of acute kidney injuries.
APE1/Ref-1 is involved in the repair of DNA damage caused by oxidative stress [19] and has been shown to possess roles in the redox regulation of various transcription factors such as activator protein-1 [20], NF-κB [21], early growth response-1 [22], NF-Y [23], HIF-1 [24], Myb [25], and p53 [26]. Moreover, this protein is involved in controlling cell-cycle arrest and apoptotic programs, with different functions in various contexts. Notably, the overexpression of APE1/Ref-1 confers resistance to apoptosis induced by chemotherapeutic drugs, radiation, hypoxia, and tumor necrosis factor (TNF) [27]. However, studies have also shown that APE1/Ref-1 is a potent signaling molecule in hyperacetylated breast cancer cells and promotes apoptosis-induced cell death [28]. In addition, H2O2 and/or hydroxyl radicals efficiently and rapidly promote a transient increase in APE1/Ref-1 protein levels, and various transcription factors are involved in the inducible expression of APE1/Ref-1 [18].
The regulatory functions modulating the activity of APE1/Ref-1 are complex and controlled through three different mechanisms [29,30]: 1) increase in APE1/Ref-1 expression after transcriptional activation; 2) re-localization of APE1/Ref-1 from the cytoplasm to the nucleus; and 3) modulation of APE1/Ref-1 posttranslational modifications such as acetylation and phosphorylation. Selenomethionine, a posttranslational modification, is another mechanism through which p53 is stimulated by an APE1/Ref-1-dependent redox mechanism [26].
Oxidative DNA damage induces apoptosis and is involved in the activation of stress response pathways, including JNK and p38 MAPK pathways [31–33]. Previous studies examining the associations of APE1/Ref-1 and MAPKs have demonstrated that these proteins have protective effects against ROS owing to the redox function of APE1/Ref-1 [34,35]. Therefore, we also hypothesized that increases in APE1/Ref-1 by H2O2-induced ROS may be a stress response to modulate the adaptation to oxidative stress. However, when APE1/Ref-1 was knocked down using siRNA, the activation of JNK and p38 decreased. The current data also revealed the transcriptional activation of NF-κB in APE1/Ref-1-overexpressing cells using promotor assays. These conclusions somewhat differed from those of most studies on APE1/Ref-1, which have evaluated cancer cells or other organs with abnormal cell signaling and survival. Moreover, these discrepancies may also be related to the use of artificial overexpression rather than the natural internal expression by ROS. Alternatively, these results may also be related to the complexity of APE1/Ref-1 signaling, which includes various mechanisms and interactions with other proteins and effector genes.
NF-κB is a major transcription factor involved in the synthesis of critical cell survival proteins in response to cellular stress [36,37]. Controversy regarding the dual roles of NF-κB in enhancing or inhibiting apoptosis has been difficult to reconcile [38]. In apoptosis, NF-κB is activated following TNF-α treatment in several cell lines [39]. Moreover, the presence of NF-κB binding sites in genes encoding interleukin-1β converting enzyme protease, c-myc, and TNF-α, which are all involved in apoptosis and cell death, has been demonstrated [1,40]. The current study revealed that H2O2 increased APE1/Ref-1 expression and apoptosis and was associated with the activation of transcription factor NF-κB.
In summary, the findings of this study demonstrated that the promoter activity of NF-κB was increased by H2O2 exposure in HK-2 cells and that this effect was blocked by siRNA targeting APE1/Ref-1. Furthermore, APE1/Ref-1 sequentially activated the ERK/p38/JNK axis in the apoptotic pathway. These findings demonstrated that APE1/Ref-1 suppression protected HK-2 cells from H2O2-induced tubular injury through the inhibition of the promoter activity of the NF-κB promoter. Therefore, APE1/Ref-1 inhibitors may represent novel therapeutic agents for the treatment of acute kidney injuries, such as I/R-induced kidney injury.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









