



Introduction
Urinary excretion of solutes, ions, and water is determined by the tubular transport removed from the glomerular filtrates. Renal tubular transport (either via reabsorption or secretion) occurs through both transcellular and paracellular pathways. Traditionally, renal regulatory function for fluid and electrolyte balance is exerted by changes in transcellular transport across the renal tubular epithelial cells. However, the regulatory roles of paracellular transport in the kidney remain incompletely known.
The junctional complexes located in the paracellular route comprise tight junctions (TJs), adherence junctions, and desmosomes [1]. The TJ is composed of three components of transmembrane bridging proteins: claudin, occludin, and junctional adhesion molecules. The C-terminus of each has a PDZ binding domain linked to scaffold zonula occludens (ZO) proteins. The ZO proteins can bind directly to cytoskeleton actin filaments [2]. In glomerular epithelial cells, the glomerular slit diaphragm has specialized transmembrane bridging proteins (nephrin and podocin) between podocyte foot processes [3].
Furuse et al. [4] identified occludin as the first component of TJs, and its abundance is related to the degree of sealing of the epithelia [5]. However, occludin knockout mice displayed well-developed TJs [6]. They also found another integral component of TJs that might have a critical role in paracellular transport. Using the same liver fraction as employed to identify occludin, a single 22-kD band was discovered by stepwise sucrose density gradient centrifugation. Peptide sequencing revealed two proteins in this band that were subsequently named claudin 1 and 2. The name “claudin” is derived from the Latin word “claudere,” which means to close [7].
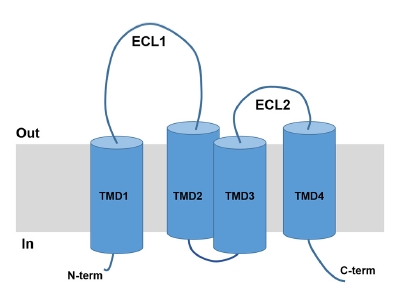
Claudins can characterize TJs because they polymerize in a linear fashion and form TJ strands with paracellular barriers or pore functions [8]. The claudin family has 27 members [9], many of which are located in the mammalian nephron [10]. Claudins contain from 21- to 28-kDa proteins and consist of four transmembrane domains, two extracellular loops (ECLs), amino- and carboxy-terminal cytoplasmic domains, and a short cytoplasmic turn (Fig. 1). The paracellular ion selectivity is determined by the charged amino acid residues located in the ECL1. The ECL2 has binding sites for claudin interactions [11]. Table 1 summarizes different claudins according to ion permeability and selectivity based on in vitro studies using cultured cell lines and ex vivo studies using knockout mice [12–42]. The results can be discrepant depending on the properties of the tested cells and animals.
Glomerular claudins
Claudins are located along with nephron segments from the glomerulus, where they exert their physiologic actions. Claudin-1 is mainly located in Bowman’s capsule or parietal epithelial cells [43]. Gong et al. [43] showed that claudin-1 overexpression was associated with overt albuminuria. In transgenic mice with claudin-1 overexpression, claudin-1 protein labeling extended to the glomerular tuft, localizing in the podocytes. Claudin-1 messenger RNA (mRNA) and protein levels also increased in the glomeruli of the representative animal model of nephrotic syndrome, puromycin aminonucleoside nephrosis (PAN). As nephrin expression declined, claudin-1 expression reached the glomerular tuft, colocalizing with nephrin in PAN glomeruli. Claudin-1 might interact with nephrin and podocin, disrupting the endogenous nephrin and podocin interactions that hold the slit diaphragm in place [43]. Thus, proteinuria can result from claudin-1 overexpression.
However, it remains unclear whether upregulation of claudin-1 is the cause of proteinuria or just a mediator of podocyte injury. In normal conditions, a high concentration of nicotinamide mononucleotide (NMN) leads to epigenetic silencing of the promoter of claudin-1 by Sirt1 in podocytes [44]. Hasegawa et al. [45] reported that, in diabetic mice, proximal tubule Sirt1 expression decreased and was followed by a decrease in NMN concentration. In the absence of NMN, the claudin-1 promoter was no longer silenced, leading to increased claudin-1 expression in podocytes and causing foot process effacement and albuminuria. Thus, proximal tubule Sirt 1 exerts regulatory action on claudin-1, but this scenario was not valid in nondiabetic animals, such as 5/6 nephrectomized mice.
Claudin-5 and claudin-6 are expressed in glomerular podocytes. When claudin mRNA levels were quantified in isolated rat glomeruli, claudin-5 expression was most abundant [46]. Its podocyte localization was demonstrated by immunoelectron microscopy and might be altered in PAN rats [46].
According to Zhao et al. [47], claudin-6 is localized in the TJs of rat podocytes. Claudin-6 was expressed in most of the tubules and glomeruli in neonates, but the expression in tubules dwindled in adults and was well-preserved in the glomeruli during development. Immunoelectron microscopy revealed that claudin-6 was distributed along the glomerular capillary wall and colocalized with ZO-1, and that its level of expression was not significantly altered in PAN rats [47].
Proximal tubule claudins
The major proximal tubule claudins are claudin-2 and claudin-10a. Claudin-2 forms paracellular channels selective for small cations such as Na+, K+, and Ca2+ and is also permeable to H2O so that 20% to 25% of proximal water absorption can occur paracellularly. It appears that cations and water travel through the same pore, where the amino acid residues in the ECL1 of claudin-2 line the narrowest part [48].
The function of claudin-2 can be inferred from knockout animals. Net transepithelial reabsorption of Na+, Cl–, and H2O was reduced from isolated perfused S2 segments of the proximal tubules in claudin-2 knockout mice [49]. These changes were associated with an increase in paracellular electrical resistance but no changes in the apical and basolateral membrane resistance that represents transcellular electrical resistance.
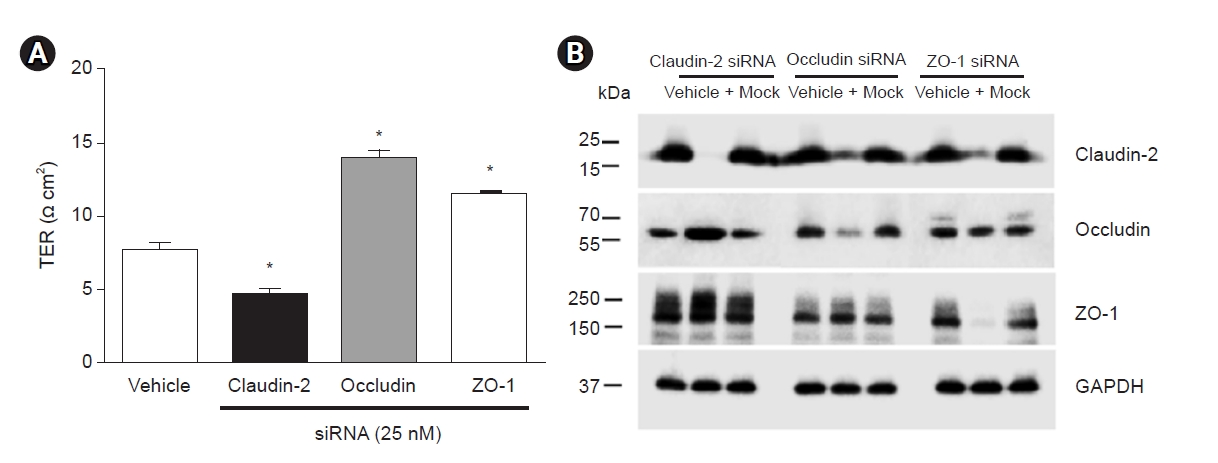
The transepithelial resistance (TER) is an indicator of permeability and varies inversely with paracellular permeability; it progressively increases from the proximal tubule or leaky epithelia to the collecting duct or tight epithelia. This finding is relevant because approximately two-thirds of the glomerular filtered fluid is reabsorbed in the proximal tubule, and fine tuning of tubular transport occurs in the distal nephron [50]. Previous claudin-2 knockdown or overexpression studies were mostly from Madin-Darby canine kidney (MDCK) or tight epithelial cells. We tested the effects of TJ protein depletion in truly leaky human kidney-2 (HK-2) cells. Fig. 2 shows TER and immunoblot results from HK-2 cells transfected with small-interfering RNAs against claudin-2, occludin, and ZO-1. With claudin-2 knockdown, an increase in occludin was associated with and might have led to a decrease in TER. When occludin was knocked down, claudin-2 was suppressed, leading to increased TER. Similarly, TER was increased by ZO-1 knockdown in association with a decrease in claudin-2 [51]. We concluded that integration of claudin-2, occludin and ZO-1 is necessary for maintaining the function of the proximal tubular epithelium.
Claudin-10 has two splice variants; -10a and -10b, respectively located in the proximal tubule and the thick ascending limb (TAL). In the proximal tubule, claudin-10a acts as an anion-selective channel (e.g., chloride absorption), whereas claudin-2 functions as a cation-selective pore [52,53]. Further independent roles of claudin-10a in the kidney remain to be determined.
The paracellular sodium transport mediated by claudin-2 contributes to energy efficiency in the kidney. Pei et al. [54] showed that claudin-2 knockout mice had larger renal oxygen consumption amounts for tubular sodium transport and a consequently lower energy efficiency. In addition, medullary hypoxia was suggested in claudin-2 knockout mice as they demonstrated remarkable furosemide-induced improvement of oxygen tension in the outer medulla. In brief, proximal tubule and TAL sodium transport are interconnected and share their load of transport. If the paracellular sodium transport is blocked in the proximal tubule, the transport load is shifted to the TAL, where Na-K-Cl cotransporter 2 (NKCC2) hyperactivity can enhance energy consumption [55].
Claudin-2 also has pathophysiological significance in calcium metabolism. Claudin-2 knockout mice demonstrate hypercalciuria due to decreased proximal tubular calcium reabsorption, which leads to papillary nephrocalcinosis and kidney stones. These results can be accentuated by decreased colonic calcium secretion or increased intestinal calcium absorption. Two large population-based studies have shown that common polymorphisms in the claudin-2 gene were associated with increased risk of kidney stones. Finally, a family case study was described in which males with a rare missense mutation in claudin-2 had marked hypercalciuria and kidney stone disease [56].
Metabolic acidosis can be associated with increased urinary calcium excretion. The protein level of claudin-2 decreased in rats with chronic metabolic acidosis and in MDCK II and HK-2 cells in response to an acidic pH [57]. The authors interpreted these results as an attempt to compensate for the chronic state of metabolic acidosis because the downregulation of claudin-2 might be associated with an increase in Na+/H+ exchanger 3 (NHE3) activity in the proximal tubule. However, Pei et al. [54] reported that the total and phosphorylated NHE3 abundance decreased by 23% and 27%, respectively, in claudin-2 knockout kidneys.
Thick ascending limb claudins
The major TAL claudins are claudin-3, -10b, -14, -16, and -19. They mediate paracellular transport of cations such as Na+, Ca2+, and Mg2+. The transcellular transport system is composed of apical NKCC2 and renal outer medullary potassium channels (ROMK) and of basolateral Na+/K+-ATPase and ClC-Kb chloride channels. In the cortex and outer stripe of the outer medulla (OSOM), the lumen-positive voltage produced by apical K+ recycling drives paracellular reabsorption of divalent cations via the claudin-16/19 complex. Here, paracellular Na+ transport can act in reverse and add to the lumen-positive transepithelial voltage. In the inner stripe of the outer medulla, however, Na+ is paracellularly reabsorbed through claudin-10b to contribute to medullary hypertonicity [58,59].
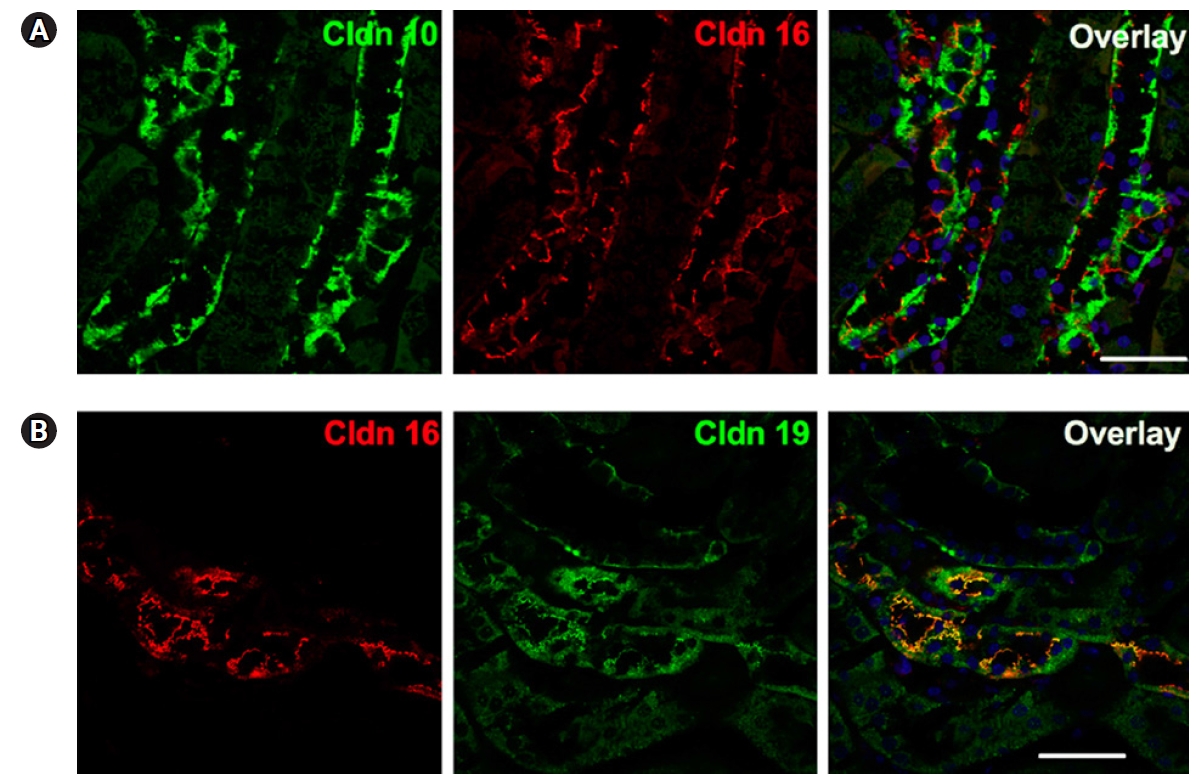
In Fig. 3, immunofluorescence microscopy shows that localization of claudin-10 does not overlap with that of claudin-16. However, claudin-16 and -19 are colocalized [60]. Similarly, claudin-3 and claudin-19 can be colocalized with each other but not with claudin-10. This characteristic claudin expression in the TAL was reported as a mosaic pattern. According to Milatz et al. [13], claudin-3 and claudin-19 were expressed in the intracellular compartment of all cortex/OSOM TAL cells. However, claudin-16 was strictly localized to the TJs. In brief, the expression of claudin-3/16/19 and claudin-10b are mutually exclusive in the TAL, and the two arrangements respectively mediate divalent and monovalent cation transport [61].
Next to the proximal tubule, the TAL is the major site of paracellular calcium transport in the kidney [61]. Divalent cations Ca2+ and Mg2+ are reabsorbed through the claudin-16/19 complex, and claudin-14 negatively regulates claudin-16 and -19 via direct interaction. During upstream signaling, microRNAs (miR-9 and miR-374) regulate claudin-14 mRNA stability and suppress translational efficacy. Gene transcription of microRNAs is regulated by the transcriptional factor nuclear factor of activated T cells (NFAT) and also via deacetylation of nearby histone molecules [62]. Consequently, claudin-14 is upregulated by stimulation of the calcium-sensing receptor (CaSR) [63].
The downregulation of claudin-2 in metabolic acidosis was described above, and we further investigated the role of TAL claudins in metabolic acidosis-induced hypercalciuria and hypermagnesiuria [64]. Fig. 4 shows that, in acid-loaded rats, both claudin-16 and claudin-19 expression decreased compared with those in controls. However, claudin-14 and CaSR expression increased in acid-loaded rats. All these changes were reversed by coadministration of the CaSR antagonist NPS-2143 and were confirmed using immunofluorescence microscopy. Hypercalciuria and hypermagnesiuria in acid-loaded rats also were significantly ameliorated by NPS-2143 coadministration. Thus, claudin-16 and claudin-19 are downregulated by metabolic acidosis via the CaSR.
Genetic defects in TAL claudins are directly linked to human diseases. The calcium- and magnesium-wasting disorder caused by either a claudin-16 or -19 mutation is called familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC). The claudin-19 disorder is accompanied by severe ocular defects and is classified as type 2 FHHNC. The phenotype of the claudin-14 mutation is characterized by deafness without renal manifestations. Claudin-10b mutations produce HELIX syndrome, which encompasses hypohidrosis, electrolyte imbalance, lacrimal gland dysfunction, ichthyosis, and xerostomia and is suggestive of abnormalities in renal ion transport, ectodermal gland homeostasis, and epidermal integrity [58].
It is interesting that claudin-14 channelopathy has no renal manifestations. However, claudin-14 knockout mice have demonstrated reduced fractional excretion of calcium and magnesium in response to high dietary calcium intake [65]. Consistent with this, claudin-14 gene polymorphisms have been associated with differences in urinary calcium excretion, whereas no associations were found with claudin-16 and -19 polymorphisms [66].
The claudin-10 mutation HELIX syndrome is characterized by lack of sweat, saliva, and tears, and it has an autosomal recessive inheritance pattern. Renal manifestations include hypokalemia, hypocalciuria, and hypermagnesemia, as shown in a case series [67]. The data from claudin-10 knockout mice can explain this renal phenotype. Conditioned knockout mice deficient in claudin-10b were generated, and the absence of claudin-10b decreased Na+ permeability and increased Mg2+ and Ca2+ permeability in isolated perfused TALs [14]. Sodium wasting might be linked to an increase in fractional excretion of potassium, and increased magnesium and calcium reabsorption could lead to hypermagnesemia and hypocalciuria in claudin-10b knockout mice. A different feature of claudin-10b knockout mice from HELIX syndrome in humans was the presence of nephrocalcinosis. Interestingly, upregulation of both claudin-16 and claudin-19 was induced in claudin-10b knockout mice and can explain these results [14].
Collecting duct claudins
Claudin-3, -4, -7, and -8 are mainly located in the collecting duct. Claudin-3 acts as a general barrier for ions, and it can promote urinary acidification due to blockage of H+ back-leak [68]. The sealing effect of claudin-3 against ions of either charge and uncharged solutes was demonstrated by its overexpression in MDCK II cells, which induced a marked increase in paracellular resistance and decreases in permeability of sodium, chloride, and larger molecules, such as 4-kDa dextran [41].
In the collecting duct, a transepithelial voltage of −25 mV with respect to the basolateral side drives Cl− transport through the paracellular channel, which is made up of claudin-4, -7, or -8 [10]. Thus, claudin-4 and -8 serve as selective anion channels, mediating a “chloride shunt,” which is coupled with transcellular Na+ reabsorption via the epithelial Na+ channel (ENaC). They may also act as Na+ barriers [68]. Collecting duct-specific knockout of either claudin-4 or claudin-8 causes hypotension, hypochloremia, metabolic alkalosis, and renal salt wasting [69,70].
Claudin-7 can form a nonselective paracellular channel that facilitates Cl– and Na+ reabsorption in the collecting duct [71]. Claudin-7 knockout mice die shortly after birth due to severe renal salt wasting and dehydration, which is suggestive of the essential roles of claudin-7 and the collecting duct paracellular NaCl transport in maintaining fluid balance [72].
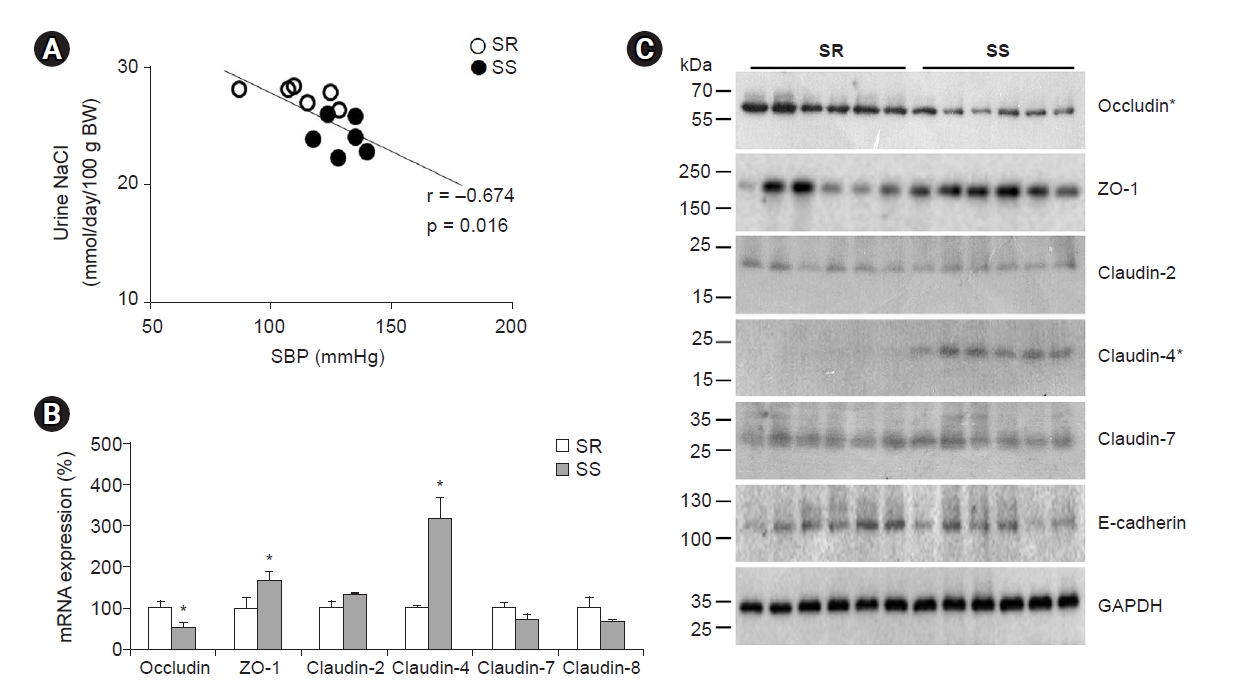
We postulated that claudin-4 or -8 upregulation contributes to salt-sensitive hypertension, and this hypothesis was tested in Dahl salt rats (Fig. 5). Compared with Dahl salt-resistant rats, Dahl salt-sensitive rats had higher blood pressure and reduced sodium excretion. In the kidney, claudin-4 protein and mRNA levels increased, and occludin protein and mRNA decreased [73]. These results might be responsible for salt retention or impaired pressure natriuresis because claudin-4 is a chloride pore, and occludin is a nonspecific or sodium barrier located along the tubule.
Hou et al. [23] reported that claudin-4 requires claudin-8 for TJ localization. Claudin-4 protein expression was suppressed by claudin-8 gene knockdown in polarized M-1 cells, whereas claudin-3 and -7 expression were not affected. In the absence of the claudin-8 gene, claudin-4 expression was confined to the endoplasmic reticulum and the Golgi apparatus and was not observed in the apical cell membrane where TJs are located.
Another regulatory factor of claudin-4 is channel-activating protease-1 (CAP1). When the cells were treated with CAP1, the expression of claudin-4 at the TJs was reduced, whereas ZO-1 expression was not affected. CAP1 decreased the cell membrane expression levels of claudin-4 and reduced paracellular Cl– permeability by disrupting claudin-4 trans-interaction [69].
In the collecting duct principal cells, aldosterone stimulates transcellular Na+ reabsorption and K+ secretion via ENaC and ROMK, respectively. In addition, aldosterone can affect paracellular Cl– absorption by regulating claudins [68]. Aldosterone activates CAP1, which inhibits claudin-4, as previously mentioned. Aldosterone also induces phosphorylation of with-no-lysine kinase-4 (WNK4), and activated WNK4 phosphorylates claudin-4 on threonine residues to promote the chloride shunt [68]. Deletion of the claudin-7 gene in collecting duct cells induced upregulation of WNK4 and ENaC [71].
We previously tested this theme in cyclosporine-treated rats because hyperchloremic metabolic acidosis is often encountered in patients using cyclosporine [74]. In the kidney, the protein expression of the Na-Cl cotransporter (NCC) was decreased by cyclosporine treatment, which suggested a decrease in transcellular chloride transport. Instead, WNK4 increased in cyclosporine-treated rat kidneys. WNK4 upregulation was confirmed by in vitro cell culture studies and in vivo immunohistochemistry [75]. However, claudin-4 phosphorylation was not demonstrated in this study.
Transcellular and paracellular transport are interlinked in the collecting duct as well. Normally, transcellular sodium absorption occurs via ENaC, and paracellular Na+ back-leak is prevented by claudin-8 barrier. When the ENaC is hyperactive, the claudin-8 barrier is strengthened to block Na+ back-leak. In contrast, when the ENaC is inactivated, the claudin-8 barrier is weakened to promote Na+ back-leak [76]. Thus, claudin-8 combines with ENaC to enable unidirectional sodium transport across the collecting duct.
Claudinopathy
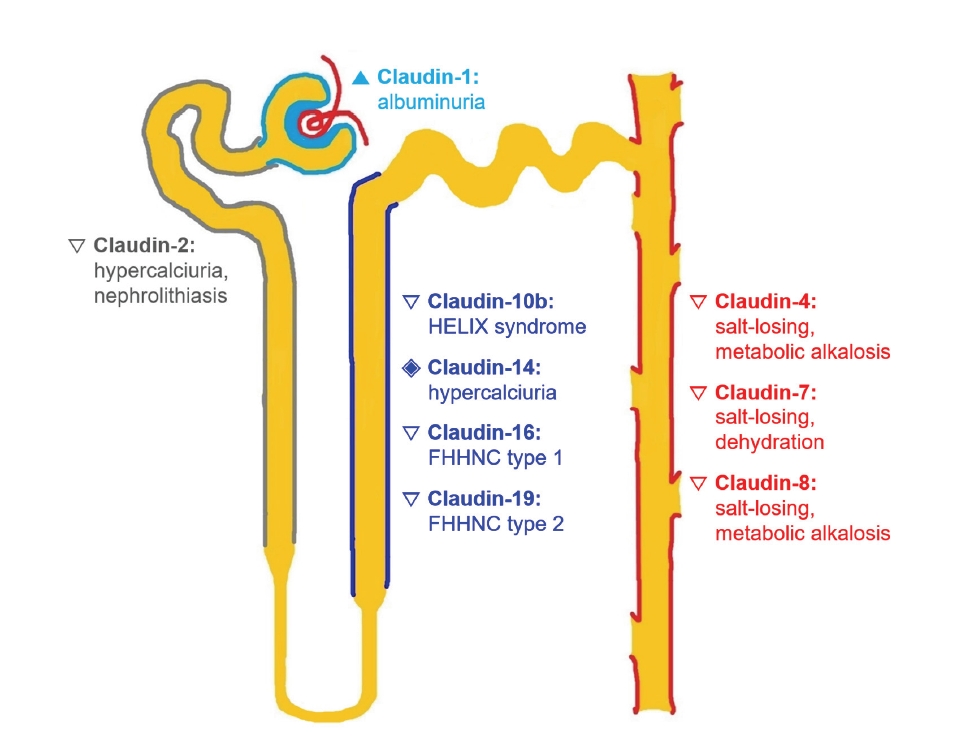
As the regulatory function and pathophysiology of claudins continue to be explored in the kidney, diseases associated with defective claudins have been termed “claudinopathies” [77]. Fig. 6 illustrates different claudinopathies along the nephron that have been described in previous experimental and clinical studies. Claudin-1 overexpression in the glomerular podocytes might have a role in albuminuria [43]. Claudin-2 and claudin-14 polymorphisms are associated with altered urine calcium excretion [56,66], which is suggestive of a role in idiopathic hypercalciuria. Hypercalciuria in metabolic acidosis is related to downregulation of both proximal tubule and TAL claudins [57,64]. Claudin-16 and claudin-19 mutations lead to FHHNC type 1 and type 2, respectively. Claudin-10b mutations can cause HELIX syndrome, which presents with hypokalemia, hypermagnesemia, and hypocalciuria [58,67]. Upregulation of claudin-4 and/or -8 may play a role in the chloride shunt, producing pseudohypoaldosteronism II and salt-sensitive hypertension [69,70].
Conclusion
Recent data from claudin studies have indicated that the paracellular pathways along the nephron are actively involved in renal physiology and pathophysiology. As the ion permeability and selectivity of different claudins continue to be defined, further studies will be required to show the regulatory and pathogenic roles of claudins in various electrolyte disorders. Understanding the interactions between paracellular and transcellular transport pathways will provide deeper insight into integrative renal physiology.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









