



Introduction
Skeletal muscle abnormalities, mostly sarcopenia, are a well-recognized phenomenon in patients with stages 3 to 5 chronic kidney disease (CKD), including those on maintenance dialysis. Extent of sarcopenia is associated with physical activity, functionality, and quality of life and represents one of the strongest predictors of mortality and hospitalization in this patient population [1,2]. Impairment of mitochondrial energy metabolism in skeletal muscle seems to play a central role in the pathogenesis of sarcopenia in patients with CKD [3]. This editorial review aims to provide insights into the growing evidence that depicts the extent of mitochondrial dysfunction in patients with moderate to advanced CKD. We include extensive metabolic, morphologic, and genomic alterations and discuss the potential therapeutic approaches of targeting mitochondrial dysfunction in the management of sarcopenia.
Mitochondrial structure and function in chronic kidney disease
The traditional methods of assessing mitochondria include muscle tissue sample examination for mitochondrial content, enzyme activity, mitochondrial protein, RNA levels and expression, and ultrastructure according to electron microscopy [3,4]. Mitochondrial abnormalities in kidney disease include decreased activity of enzymes such as citrate synthase and hydroxyl acyl CoA dehydrogenases and abnormal mitochondrial ultrastructure (Table 1). Specifically, studies on muscle biopsies from patients on maintenance hemodialysis (MHD) revealed various abnormalities in skeletal muscle mitochondria including signs of fragmentation [5], cristae swelling [6,7], presence of autophagosomes [5], and increased lipofuscin pigment, indicative of oxidative damage [7]. Some studies suggest the presence of increased mitophagy, selective removal of mitochondria by autophagy, in patients with CKD [5,7]. Mitochondrial dynamics (i.e., fusion and fission of mitochondria), which are important for proper mitochondrial function, can be altered in patients with moderate to advanced CKD. Some studies have found increased mitochondrial fission in skeletal muscles of such patients. These data suggest that damaged mitochondria in CKD can be segregated by mitochondrial fission and removed by mitophagy.
Various murine CKD models as well as human studies have shown that decreased muscle mitochondrial content is associated with reduced physical performance [3]. Furthermore, in animal models of CKD, it has been shown that reduction in mitochondrial function precedes changes in muscle mass and muscle strength [3]. A recent study in humans showed a molecular signature of mitochondrial dysfunction in age-related sarcopenia similar to that in kidney disease [8]. Noninvasive techniques such as (31)P-magnetic resonance spectroscopy (31P MRS) allow evaluation of mitochondrial dysfunction through measurement of phosphocreatine recovery, which depends on mitochondrial oxidative phosphorylation [9]. Studies based on these assessment techniques have shown that mitochondrial dysfunction is present in patients with CKD and worsens as kidney function declines. The mitochondrial function also correlates with physical performance, muscle quality, systemic inflammation, and burden of oxidative stress, highlighting the complex pathophysiology of mitochondrial dysfunction in CKD.
While mitochondrial dysfunction has been studied mostly in skeletal muscle, there is evidence for disrupted mitochondrial function in the nervous and cardiovascular systems. In animal models, CKD has been associated with dysfunction of mitochondrial complexes I and II in various regions of the brain, including cortex, striatum, and hippocampus [10], and might be related to the cognitive decline seen in CKD [11]. Similarly, CKD is associated with disruption of mitochondrial structure in cardiac tissue in rat models [12]. Mitochondrial function is of paramount importance in the heart due to its high energy requirement and mitochondrial dysfunction and has been linked with many cardiovascular diseases including cardiomyopathies, heart failure [13], and cardiorenal syndrome [12]. Cardiac tissue from animal models of CKD showed swollen mitochondria, decreased mitochondrial volume, disrupted cristae [12], and downregulated ubiquinone biosynthesis [14].
Mechanisms of mitochondrial dysfunction in patients with kidney disease
The mechanisms underlying mitochondrial dysfunction in the setting of moderate to advanced kidney disease are complicated and likely multifactorial (Fig. 1). Data are implicating the impairment of an array of mitochondrial enzymes in the development of mitochondrial dysfunction in CKD. Diminished activities of mitochondrial electron transport chain (ETC) complexes and pyruvate dehydrogenase are shown in murine models of early CKD before any detectable decline in muscle power [3]. Defects in the switch from anaerobic to aerobic metabolism caused by ineffective pyruvate dehydrogenase are shown to induce lactate accumulation and muscle fatigue commonly found in CKD patients [3]. Furthermore, tissue samples from patients on maintenance dialysis demonstrated decreased protein expression of mitochondrial import pores associated with mitochondrial membrane potential and cytochrome C oxidase activity [4]. Decreased activity of citrate synthase and cytochrome c oxidase and slower synthetic rates of muscle mitochondrial proteins have been found in biopsy samples from patients with stages 3 and 4 CKD [15]. A potential explanation for suppressed mitochondrial biogenesis is overall decrease in synthesis of muscle proteins in the state of protein-energy wasting associated with advanced kidney disease [3,15,16].
Studies of plasma metabolomics also revealed several distinct disturbances in mitochondrial metabolism of patients with advanced CKD. Available data suggest notable differences in the tricarboxylic acid cycle and deficiency of metabolites involved in coenzyme Q (CoQ, also known as ubiquinone) biosynthesis, a crucial component of mitochondrial ETC. Furthermore, defects in nicotinamide metabolism and nicotinamide adenine dinucleotide synthesis through impaired tryptophan metabolism were identified [17]. These changes can compromise the efficiency of oxidative phosphorylation and result in the uncoupling of oxidative phosphorylation in the setting of kidney disease [7,18]. Furthermore, CoQ10 deficiency, as shown in CKD patients [19], is known to trigger the generation of superoxide during electron transport in the mitochondria [20], leading to further oxidative damage.
Another energy pathway compromised by CKD is the beta-oxidation of fatty acids, a major energy source during the fasting state. Defective beta-oxidation is characterized by accumulation of intermediate chain acylcarnitines and decreased long-to-intermediate acylcarnitine ratio [21]. Lipidomic profiling of patients with stages 2 to 5 CKD showed a graded decrease in the long-to-intermediate acylcarnitine ratio with increasing severity of kidney disease, leading to impaired beta-oxidation. Importantly, the lipid deposits caused by ineffective beta-oxidation are trapped in the mitochondrial matrix and undergo lipid peroxidation by reactive oxygen species (ROS), promoting further mitochondrial damage and creating a cycle that results in cellular death [22,23]. Lipotoxicity due to downregulated beta-oxidation is a well-recognized abnormality in multiple glomerular diseases [22,24,25] and might have a critical role in muscle mitochondrial dysfunction in CKD that should be investigated in future studies.
CKD patients have been found to harbor significant mitochondrial DNA (mtDNA) damage. Similar to age-related sarcopenia, muscle loss in CKD appears to be associated with mtDNA deletions [26], which correlate negatively with complex I activity [27]. Indeed, analysis of patients from the HEMO study found that one-third of patients had an mtDNA 4977-bp deletion that was predictive of poor survival. The mtDNA copy number was also a predictor of survival because a higher copy number, indicating higher mitochondrial content, was associated with better outcomes among patients on MHD [26].
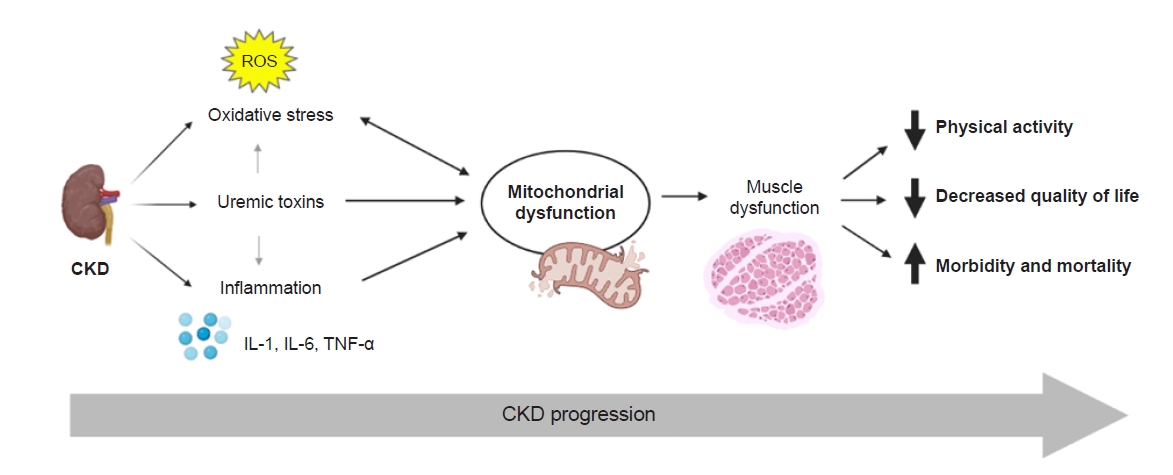
Possible mechanisms by which kidney disease could induce mitochondrial damage include uremic toxins, oxidative stress, and inflammatory milieu (Fig. 2). In murine models, uremic toxins including indoxyl sulfate, p-cresol sulfate, indole-3-acetic-acid, l-kynurenine, and kynurenic acid impair mitochondrial function by inducing mitochondrial fission [28], autophagy [28], and disrupting the activities of complex III and IV enzymes in the electron transport system [29]. Similarly, in vitro exposure of muscle cells to uremic serum suppresses mitochondrial oxidative capacity [4]. Evidence suggests that CKD patients are affected by increased levels of oxidative stress from the early stages of the disease due to increased ROS production and antioxidant depletion [30], with progressive CKD leading to a worse prooxidant state [31]. As a source of oxidative reactions, mitochondria are susceptible to oxidative damage. ROS directly induce mitochondrial uncoupling [32], as seen in patients with stages 3 and 4 CKD [18]. Additionally, skeletal muscle tissue from patients on MHD shows evidence of enhanced oxidative damage to mitochondrial proteins, lipids, and DNA along with somatic mtDNA mutations [33]. Oxidative damage to mitochondrial components triggers mitophagy, shifting the mitochondrial dynamic toward increased mitochondrial removal and reduced mitochondrial mass [34], which is a key finding in CKD-induced myopathy [5,7,28]. We have shown that the ratio of isofurans to F2-isoprostane, an indicator of oxidative stress, is associated with mtDNA copy number and mitochondrial volume density as well as severity of CKD [7]. These data suggest direct correlation between CKD-induced mitochondrial dysfunction and oxidative stress and the ratio of plasma isofurans to F2-isoprostane as a potential biomarker for mitochondrial dysfunction in CKD [7].
Finally, proinflammatory cytokines, such as interleukin (IL)-6 and IL-1, have been implicated in the development of sarcopenia in patients on MHD [35] as well as in other chronic inflammatory states [36,37]. Tumor necrosis factor (TNF)-α and IL-6 were found to suppress inducers of mitochondrial biogenesis and augment inducers of mitophagy in murine CKD models [3], while TNF-α and IL-6 inhibitors effectively attenuated these changes [3]. In muscle cell models, exposure to increasing concentrations of TNF-α and IL-6 correlated with decreasing mitochondrial activity, which could be reversed partially by the anti-inflammatory adipokine adiponectin.
Mitochondrial function and physical performance in kidney disease
A clinically relevant effect of skeletal muscle mitochondrial dysfunction is its potential impact on physical activity and performance. A study of 12 nondialysis-dependent CKD patients showed that lower eGFR was associated with decreased mean energy coupling and greater O2 uptake. Importantly, CKD patients in the study had preserved physical performance [18], suggesting that development of mitochondrial dysfunction preceded initiation of maintenance dialysis and onset of clinically significant functional decline. Corroborating these findings [18], Kestenbaum et al. [38] showed that the presence and severity of CKD negatively correlated with muscle mitochondrial function measured by 31P MRS in lower leg muscles of 53 patients with stage 3 and 4 CKD. Two independent studies also showed that mitochondrial function correlated with 6-minute walk performance, suggesting a direct link between mitochondrial function and exercise tolerance in patients with CKD [38,39].
Therapeutic approaches
The recent understanding of CKD-associated mitochondrial dysfunction has implications for potential therapeutic approaches to provide mitochondrial protection and maintain or improve functional capacity in patients with kidney disease. Among the wide variety of approaches to modulate mitochondrial function [40], CoQ10 is an important target since CKD patients are shown to have CoQ10 deficiency [19]. CoQ10 supplementation has been used successfully to target mitochondrial dysfunction in heart failure patients [41] and showed beneficial effects on the metabolic profile in CKD patients [42]. There are ongoing clinical trials designed to assess the effects of CoQ10 supplementation and nicotinamide riboside supplementation on mitochondrial energetics and exercise capacity in the CKD population (ClinicalTrials.gov NCT03579693).
Cardiolipin, an inner mitochondrial membrane phospholipid that facilitates electron transfer by cristae modulation, has been studied as a candidate target [43]. Elamipretide (SS-31), which selectively binds cardiolipin and inhibits cardiolipin peroxidation during oxidative stress [44], was shown to have renocardioprotective properties [45,46] and to improve skeletal muscle mitochondrial energetics [47–49] in animal models. SS-31 restored age-related mitochondrial dysfunction including uncoupling of oxidative phosphorylation, decline in maximal adenosine triphosphate (ATP) production, and increasing ROS production while increasing exercise endurance capacity [47]. Additional studies are required to determine its utility in CKD-associated mitochondrial dysfunction. A number of other novel mitochondria-targeted therapies, including mitochonic acid 5, carnitine supplements, activators of AMP kinase, and peroxisome proliferator-activated receptor-gamma coactivator 1α, has produced promising results for treatment of glomerular disease [50] and should be studied for treatment of CKD-related sarcopenia.
Exercise is known to improve mitochondrial function in the general population. However, the beneficial effects of aerobic training, such as improvements in muscle quality and quantity, strength, and physical functioning, are not observed consistently in patients with advanced kidney disease. In healthy individuals, possible positive effects of exercises on muscle can be partly explained by PGC1-α, a transcriptional coactivator that activates mitochondrial biogenesis and peroxisome proliferator-activated receptor-α. PGC1-α is highly abundant in skeletal muscle and its expression is activated by aerobic training. PGC1-α-related effects of exercise on muscle have been demonstrated in transgenic mouse models. With aging, expression of PGC1-α is suppressed. The lack of effect of exercise on skeletal muscle in patients with advanced kidney disease might be related to reduced expression of PGC1-α, which is mandatory for proper mitochondrial function. A recent study has shown that 12 weeks of exercise training does not increase mitochondrial content despite an increase in PGC1-α [51]. This finding suggests downstream inhibition of the PGC1-α pathway. A possible explanation is activation of the factor forkhead box O1 (Foxo1), which induces skeletal muscle atrophy in several disease states including CKD [52]. Foxo1 also impairs mitochondrial function and inhibits mitochondrial biogenesis through inhibition (by acetylation) of PGC1-α [53]. Further studies should evaluate an appropriate exercise training for patients with CKD or combination of exercise and mitochondrial-targeted interventions to improve the response to exercise.
Despite the potential of the above strategies to improve mitochondrial function in disease states, there are important barriers in identifying the most relevant mitochondrial targets and effective drug delivery methods. Drug safety continues to be a major concern, especially for antioxidant interventions since high doses are needed for efficacy, and molecular delivery inside mitochondria is difficult to achieve in high concentrations [54]. While many novel therapeutic options are being introduced at the preclinical level, clinical trials are limited.
Summary
Abnormalities in skeletal muscle structure and function are observed commonly in patients with kidney disease and worsen as kidney function declines. Mitochondrial dysfunction, presenting with a variety of morphological and functional changes that compromise ATP production, has a major role in the development of muscle wasting from the early stages of CKD and is a potential marker of increasing disease severity. While the exact pathophysiological mechanisms remain to be identified, accumulation of uremic toxins, enhanced oxidative stress, and systemic or local inflammation could lead to mitochondrial damage. Understanding these pathways could pave the way in the development of mitochondria-targeted therapies for muscle wasting in patients with moderate to advanced CKD.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









