Fructose in the kidney: from physiology to pathology
Article information

Abstract
The Warburg effect is a unique property of cancer cells, in which glycolysis is activated instead of mitochondrial respiration despite oxygen availability. However, recent studies found that the Warburg effect also mediates non-cancer disorders, including kidney disease. Currently, diabetes or glucose has been postulated to mediate the Warburg effect in the kidney, but it is of importance that the Warburg effect can be induced under nondiabetic conditions. Fructose is endogenously produced in several organs, including the kidney, under both physiological and pathological conditions. In the kidney, fructose is predominantly metabolized in the proximal tubules; under normal physiologic conditions, fructose is utilized as a substrate for gluconeogenesis and contributes to maintain systemic glucose concentration under starvation conditions. However, when present in excess, fructose likely becomes deleterious, possibly due in part to excessive uric acid, which is a by-product of fructose metabolism. A potential mechanism is that uric acid suppresses aconitase in the Krebs cycle and therefore reduces mitochondrial oxidation. Consequently, fructose favors glycolysis over mitochondrial respiration, a process that is similar to the Warburg effect in cancer cells. Activation of glycolysis also links to several side pathways, including the pentose phosphate pathway, hexosamine pathway, and lipid synthesis, to provide biosynthetic precursors as fuel for renal inflammation and fibrosis. We now hypothesize that fructose could be the mediator for the Warburg effect in the kidney and a potential mechanism for chronic kidney disease.
Introduction
Fructose is a natural sugar present in fruits and honey and is a fundamental nutrient for wild animals. Bears, squirrels, birds migrating over long distances, and freshwater Pacu fish actively eat fruits to accumulate fat, presumably as a protection against periods of food shortage [1]. Fructose contributes to lipid and glycogen syntheses for energy storage, and to the development of insulin resistance to prevent glucose utilization in the peripheral tissue, and glucose delivery to the central nervous system. In addition, fructose also stimulates salt reabsorption to raise blood pressure (as discussed in the following section) [2]. Fructose is also metabolized under hypoxic conditions and often exhibits the protective effect. A recent study examined how the naked mole rat could survive long periods of hypoxic conditions and found that it was attributed to their ability to produce fructose endogenously in several organs, which is subsequently metabolized to provide several biosynthetic precursors required for cell survival, including nucleic acids, amino acids, lipids and energy [3]. Likewise, the reason why the fetus exposed to hypoxia can survive during early pregnancy is that the developing placenta also produces endogenous fructose, likely aiding fetal organ growth in wild animals as well as humans [4–6].
In modern society, fructose, as a component of high-fructose corn syrup or table sugar, is preferentially added to sugar-sweetened soft drinks and sodas. A dramatic increase in fructose consumption is, however, associated with a high prevalence of the metabolic syndrome, stimulating a heated debate over the potential danger of sugar-sweetened beverages (SSB) [7,8]. Likewise, several clinical studies have sought the role of fructose in the kidney, but the issue remains controversial. Interestingly, more than two SSB per day is associated with an incidence and a prevalence of chronic kidney disease (CKD) [9,10], but less than one SSB per day was not [11,12]. Fructose may therefore impair renal function in a dose-dependent manner. An intervention study also showed that a low-fructose diet lowered blood pressure and reduced systemic inflammation in subjects with CKD [13].
A recent scientific discovery is that fructose is produced endogenously, and is involved in the pathogenesis of several types of disorders. Acute kidney injury, diabetic nephropathy, cardiac hypertrophy, aging, and salt-sensitive hypertension are now recognized to be mediated by endogenous fructose.
This article summarizes the basis of fructose physiology, discusses a potential mechanism by which fructose causes kidney disease, and finally proposes our hypothesis that fructose mediates the Warburg effect in CKD.
Current concepts and update regarding dietary fructose metabolism
It has long been assumed that the liver is the primary site for dietary fructose metabolism, but recent studies have demonstrated that the small intestine plays a substantial role in dietary fructose metabolism [14]. After sucrose is digested by sucrase into fructose and glucose, fructose is absorbed by enterocytes via the glucose transporter (GLUT) 5 at the apical membrane of enterocytes. Since the intestine contributes ~25% of systemic gluconeogenesis both after prolonged fasting and in diabetes [15], intestinal epithelium likely utilizes dietary fructose as a substrate for gluconeogenesis (Fig. 1). However, when present in excess, fructose saturates the intestinal metabolic capacity. Excessive fructose either spills over to the colon or is transported through the GLUT2 from the basal membrane into the portal vein and then to the liver [14]. Interestingly, intestinal fructose metabolism determines an individual’s preference for sweet tastes and sugar intake but does not contribute to the development of metabolic syndrome [16]. In the colon, fructose is likely digested by microbiota that use fructose carbons to generate tricarboxylic acid (TCA) cycle intermediates, essential amino acids, and short-chain fatty acids [14]. In turn, fructose spilling over from intestinal shield acts on the hepatocyte via GLUT2 and drives the metabolic syndrome [16]. Hepatic fructose metabolism is associated with increased hepatic fatty acid and malonyl-CoA synthesis, reduced fatty acid oxidation, and modification of the mitochondrial proteome [17]. Similar to the enterocyte, excessive fructose in the kidney likely escapes into the systemic circulation. In the kidney, fructose in systemic circulation is filtered through the glomerulus into the urinary space, and urinary fructose is reabsorbed by the proximal tubular cells (Fig. 1).
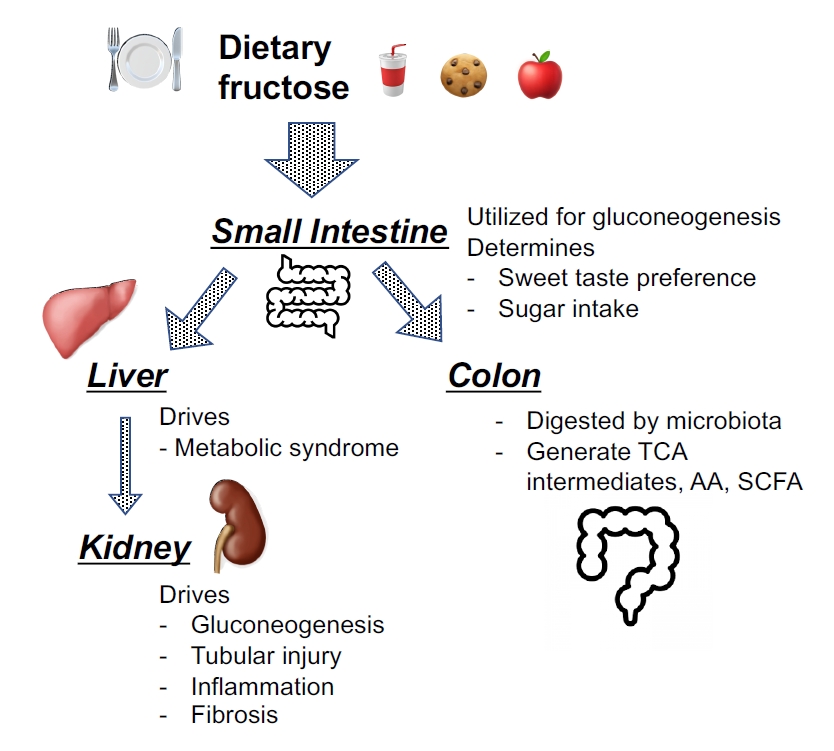
Current concepts regarding the metabolic pathways of dietary fructose.
Dietary fructose is absorbed by enterocytes via glucose transporter (GLUT) 5 at the apical membrane of enterocytes and is likely utilized as a substrate for gluconeogenesis. Interestingly, intestinal fructose metabolism determines an individual’s preference for sweet tastes and sugar intake but does not contribute to the development of the metabolic syndrome. When present in excess, fructose may saturate intestinal metabolic capacity and spill over to the colon where fructose can be digested by gut microbiota and utilized for the generation of tricarboxylic acid (TCA) intermediates, essential amino acids (AA), and short-chain fatty acids (SCFA). Alternatively, fructose exceeding the metabolic capacity of enterocytes is excreted from GLUT2 at the basal membrane and passes into the portal circulation, and reaches the hepatocytes. The fructose is then reabsorbed via GLUT2 at the surface of hepatocyte to be metabolized, likely driving the metabolic syndrome. Hepatic fructose metabolism is associated with increased hepatic fatty acid synthesis and malonyl-CoA levels, and a reduction in fatty acid oxidation. Similar to the enterocyte, excessive fructose likely escapes from hepatocyte into the systemic circulation. The kidney also plays a role in reabsorption and excretion of fructose. At physiological concentrations, fructose is utilized for gluconeogenesis, whereas it causes kidney injury when present in excess.
Fructose transporters are predominantly expressed in the proximal tubules
After fructose is metabolized in the liver, only small amount of fructose escapes from the liver to reach the systemic circulation, and therefore serum fructose concentrations range from 0.1 to 0.8 mM [18]. After filtration through the glomerulus, urinary fructose is either reabsorbed in the proximal tubular epithelial cells (Fig. 2), or excreted in the urine.
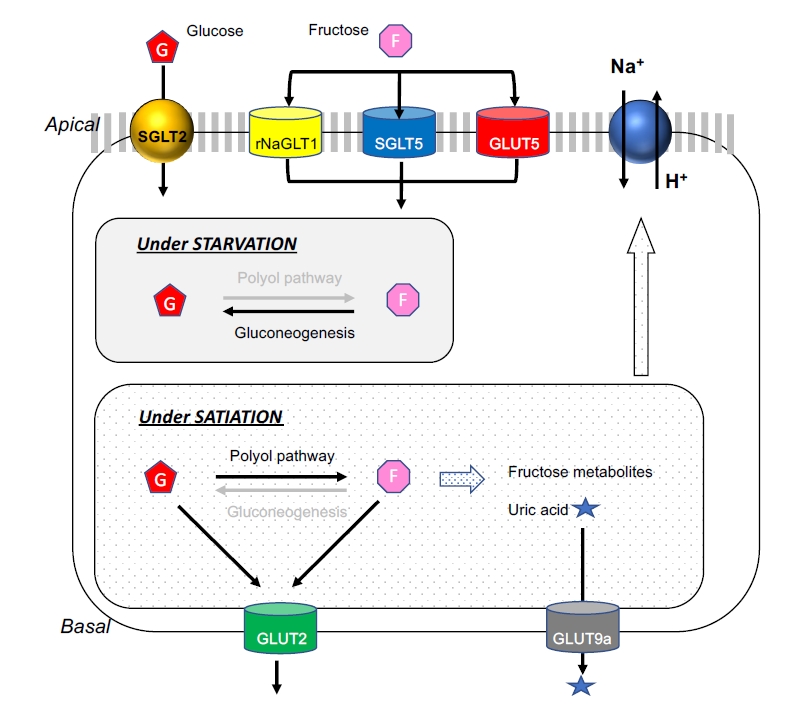
Fructose transporters and metabolism in the proximal tubules.
In the proximal tubules, urinary fructose is reabsorbed at the apical membrane of the epithelial cells via several types of fructose transporters. The glucose transporter (GLUT) 5 is considered to be a major transporter for fructose. The sodium glucose cotransporter 5 (SGLT5) is a high-affinity kidney-specific transporter for fructose and mannose in humans and mice, while the rat sodium-dependent glucose transporter-1 (rNaGLT1) is also expressed in both convoluted and straight proximal tubules in the rat and also mediates fructose transport. Under physiological conditions or during starvation, fructose is utilized as a substrate for gluconeogenesis. In turn, during satiation or when fructose is in excess, fructose is metabolized in the cytosol to produce several fructose metabolites, including uric acid. The GLUT2 facilitative transporter is expressed in the basal membrane. When fructose concentrations are higher in the cytosol than in the blood of peritubular capillary, GLUT2 transports intracellular fructose into the blood in the peritubular capillary. Likewise, GLUT9a, another facilitative transporter, is expressed in the basolateral membrane and favors urate transport back into the circulation from the tubular cells.
GLUT5, a high-affinity facilitative transporter, is considered to play a major role in fructose transport and is expressed at the apical membrane of the epithelial cells in the straight portion of the proximal tubule [19,20]. An alternate fructose transporter is the sodium glucose cotransporter 5 (SGLT5), which is a high-affinity transporter for fructose and mannose in humans and mice [21,22]. This transporter is exclusively expressed in the kidney, and likely located in the S2 segment of the proximal tubular cells [23]. The rat sodium-dependent glucose transporter 1 (rNaGLT1) is also expressed at the apical membrane of epithelial cells in both the convoluted and straight proximal tubules in the rat, and also mediates fructose transport [24]. While GLUT9 (SLC2A9) is a member of the facilitative GLUT gene family, it is now primarily described as a urate transporter (URAT) that can exchange both fructose and glucose for urate. The two splice variants of GLUT9, GLUT9a (full length) and GLUT9b (ΔN) are both present in the human kidney [25]. The GLUT9a (540 amino acids) splice variant is expressed in the basolateral membrane of the proximal tubular epithelial cells and favors urate transport back into the circulation from the tubular cells [26]. In turn, the GLUT9b (512 amino acids) splice variant is expressed at the apical site, and likely transfers urate from tubular fluid into cells [26,27] and the collecting ducts [25,28] in humans.
Alternatively, GLUT2 may transport fructose from the basolateral membrane of the proximal tubular cells into the systemic circulation [23,29], perhaps when fructose is abundant in the cytosol (Fig. 2). Given GLUT2 is a facilitative transporter operated by a passive diffusion process, it may act to excrete fructose when the intracellular fructose concentration is greater than that of the blood.
Physiology of fructose metabolism in the proximal tubules
The straight segment of the proximal tubules exclusively expresses GLUT5 so that it may be the primary site for fructose metabolism. However, both fructokinase and aldolase B, another key enzyme for fructose metabolism, are also present in the convoluted proximal tubules [30,31], suggesting that fructose metabolism is not restricted in the straight segment, but is also likely operated in the convoluted segment of renal tubules.
Fructose are likely utilized as substrates for gluconeogenesis in the proximal tubules, where gluconeogenesis is dominant over glycolysis. In fact, several gluconeogenesis enzymes, including phosphoenolpyruvate carboxykinase, fructose bisphosphatase, and enzymes of the glucose 6-phosphatase system are dominantly activated [32,33], while glycolysis enzymes are less activated [34–36] in the proximal tubules compared to other parts of nephron.
In 1961, by utilizing in situ perfusion in the rat, Salomon et al. [37] directly measured the difference between arteriovenous fructose and glucose concentrations after bolus infusion of 25 mg of fructose into the peripheral vessels. The reduction of fructose concentrations after passage of blood through the kidney was associated with equivalent increases in the renal venous glucose; the extent of fructose disappearance and the appearance of glucose averaged approximately 19%. In 1982, Björkman and Felig [38] found that intravenous infusion of fructose in humans at 2 mmol/min for 135 minutes resulted in a rise in glucose concentration in the renal vein (0.17 ± 0.05 mmol/L). The results indicated that 20% of intravenously infused fructose was taken up by the kidney, and the net glucose release from the kidney could be derived from 55% of the net renal uptake of fructose.
Proximal tubular cells go wrong with excessive fructose
When the proximal tubular cells are overloaded with excessive fructose after satiation, fructose metabolism likely becomes dysregulated and causes pathological reactions (Fig. 2, 3). In experimental studies, normal rats developed mild tubulointerstitial injury with inflammation and fibrosis when fed a high-fructose diet [19,39]. In the case of preexisting kidney injury, fructose accelerates tubular injury and interstitial inflammation and fibrosis in CKD rats [40]. An in vitro study showed that the cultured proximal tubular cells released inflammatory cytokines, including monocyte chemoattractant protein-1, in response to pathological fructose concentrations; interestingly, this reaction was found to be mediated by uric acid [41].
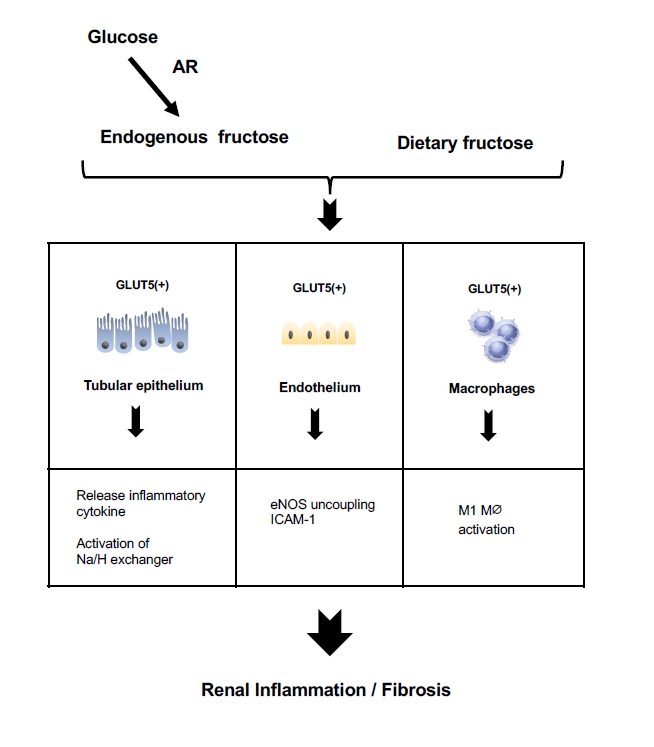
Postulate mechanism of fructose-induced kidney disease.
Fructose, arising either from the diet or from endogenous production under pathological conditions, acts on the tubular epithelial cells, endothelial cells, and macrophages through fructose transporters, such as glucose transporter 5 (GLUT5), to cause inflammation and fibrosis in the kidney.
AR, aldose reductase; eNOS, endothelial NO synthase; ICAM-1, intercellular adhesion molecule-1.
Endothelial cells express GLUT5 [42] and release intercellular adhesion molecule-1 (ICAM-1) in response to fructose [43]. A likely mechanism for this effect is the ability of fructose to reduce nitric oxide (NO) availability due to uncoupling of endothelia nitric oxide synthase (eNOS), as NO donors mitigated the fructose-induced ICAM-1 expression [43–45]. Fructose-induced generation of uric acid could be also involved in this process, as uric acid directly impairs endothelial function [46,47].
Likewise, macrophages are also a target for fructose given they express GLUT5 (Fig. 3). Several experimental studies have shown that a high-fructose diet causes renal inflammation with macrophage infiltration in rodents [19,40,43]. In vitro studies confirmed the ability of fructose to act directly on macrophages via GLUT5 to release inflammatory cytokines [48,49]. Macrophages exhibit two phenotypes, a proinflammatory M1 and an anti-inflammatory M2 phenotype; fructose is likely an ideal fuel for M1 macrophage, which relies on glycolysis. However, the mechanism by which fructose activates macrophages may be somewhat complex. A recent study of fructose-induced inflammation demonstrated that oxidative metabolism, but not glycolysis, plays a dominant role in macrophage activation [48]. While a basic concept is that uric acid suppresses aconitase in the TCA cycle and hence reduces mitochondrial respiration, the investigators identified an alternative pathway for fructose to stimulate the TCA cycles. They found that glutamine was incorporated into the TCA cycles in response to fructose, and supplies α-ketoglutarate that can bypass this step allowing oxidative phosphorylation to occur in human monocytes and mouse macrophages. Therefore, macrophages likely utilize either glycolysis or oxidative phosphorylation, perhaps depending on cellular conditions. While it remains unclear how macrophages switch these pathways, a key trigger may be oxygen availability. Under severe hypoxia, cytochrome c oxidase activity decreases [50], and glycolysis is dominant [51]. In contrast, under aerobic conditions, cytochrome c activity is activated, and glycolysis is completely replaced by oxidative phosphorylation [51].
Fructose causes salt-sensitive hypertension
Fructose intake is likely associated with hypertension in humans [52]. Experimental studies have shown that rats fed a high-fructose diet had elevated blood pressure in response to additional salt intake (Fig. 2). The proximal tubules play a key role in salt handling, as the majority of Na+ filtered through glomerulus is reabsorbed into the tubular epithelial cells via Na+/H+ exchangers (NHEs) located in the apical membrane [53,54]. Fructose-induced salt sensitivity can be accounted for by the ability of fructose to stimulate both the expression and the activity of NHEs, and increase Na+ reabsorption in the proximal tubules [53]. While NHEs are regulated by angiotensin II, fructose sensitizes the proximal tubules to angiotensin II by upregulating NHE expression [53,54]. In addition, urate also plays a key role in the development of salt-sensitive hypertension in response to fructose as it causes arteriolopathy, tubulointerstitial injury, and a reduction in NO in endothelial cells [43,46,47,55].
Renal proximal tubular cells turn on glycolysis when injured
The proximal tubular cells normally prefer lipids over glucose for energy generation, so glycolysis is not operative in this cell type. It is because enzymes for gluconeogenesis are dominantly activated over glycolytic enzymes; and therefore, fructose metabolism is physiologically linked with gluconeogenesis, but not with glycolysis [56]. However, this is unlikely the case when the tubular cells are damaged. In fact, damaged proximal tubular cells are often associated with mitochondrial alteration, resulting in a metabolic switch from mitochondrial oxidative phosphorylation to glycolysis with amplified expression of glycolytic enzymes [57]. Thus, both fructose and glucose are metabolized in damaged proximal tubular cells.
Does the combination of fructose with glucose accelerate glycolysis in renal proximal tubular epithelial cells?
The combination of fructose with glucose modifies the activation of glucokinase, the enzyme that catalyzes the first step of glycolysis. In hepatocytes, glucokinase is positively regulated by fructose 1-phosphate (Fru1P) whereas it is inhibited by fructose 6-phosphate (Fru6P) [58,59]. The mechanism for Fru1P-mediated glucokinase activation is the release of glucokinase from glucokinase regulatory protein (GKRP), which sequesters glucokinase in the nucleus [60,61]. Even at low concentrations, intracellular fructose is rapidly metabolized to Fru1P. Therefore, Fru1P-induced glucokinase activation may explain how fructose facilitates glucose utilization. Consistent with these findings, Shiota et al. [62] showed that small amounts of fructose enhanced hepatic glucose uptake in the dog. Furthermore, fructose metabolism also increases fructokinase activity, which depletes intracellular adenosine triphosphate (ATP). Since ATP negatively regulates the glycolytic pathway by inhibiting phosphofructokinase and pyruvate kinase, the ATP depletion due to fructokinase activation enhances glycolysis. However, these processes may not occur in the kidney, given glucokinase (hexokinase IV) is expressed only in hepatocytes and pancreatic β cells, [63] while renal proximal tubular cells express hexokinase I and II [57,64].
Endogenous fructose may be a unifying pathway in the development of chronic kidney diseases
Interestingly, fructose is produced endogenously in the kidney, particularly under condition of ischemia/hypoxia, high osmotic stress, aging, pressure overload, and diabetes. A potential mechanism for fructose synthesis is the activation of the polyol pathway, in which glucose is reduced by aldose reductase to sorbitol, which is then oxidized by sorbitol dehydrogenase to fructose. Therefore, fructose can be readily produced when glucose is constantly supplied. A key step in this process is the activation of aldose reductase, which can be stimulated by several factors, including hypoxia, osmotic stress, and diabetes, and may also account for the findings that several factors induce endogenous fructose production in several pathological conditions.
Cardiac surgery often results in postoperative acute kidney injury due to ischemia. Our research group studied pediatric patients who underwent cardiac bypass surgery and found that urinary fructose concentrations were elevated in patients with ischemic acute kidney injury (iAKI) compared with patients without iAKI [65]. Mice with iAKI also exhibited increased renal fructose concentrations [65], suggesting that ischemia could stimulate endogenous production of fructose in the kidney. Similarly, compared with nondiabetic mice, diabetic mice also had higher fructose levels in the kidney due to the activation of the polyol pathway [66]. Importantly, both studies demonstrated that blocking fructose metabolism ameliorated tubular injury induced by either ischemia or diabetes in mice lacking the fructokinase gene [65,66].
In the senescent kidney, endogenous fructose production likely contributes to the development of glomerular injury. A mouse study showed that glomerular injury accompanied by glomerular hypertrophy, collagen IV deposition, and mesangiolysis was observed in aging wild-type mice while aging fructokinase-knockout mice developed significantly less glomerular injury [67]. In turn, high salt intake also stimulates endogenous fructose production in the liver, while blocking fructokinase slows fat accumulation in the epididymis [68]. In addition, in the mouse heart, pressure-overload-induced cardiac hypertrophy was ameliorated by blocking fructose metabolism [69].
Pathways downstream from fructose metabolism may contribute to kidney disease
In the kidney, inflammation and fibrosis are accompanied with several pathological steps, including cell proliferation, enzymatic activation, and protein synthesis, which require several biological factors, including energy sources, nucleotides, lipids, and redox balance, which can be efficiently provided by aberrant glycolysis. Fructose is metabolized through several pathways and contributes to the progression of CKD (Fig. 4).
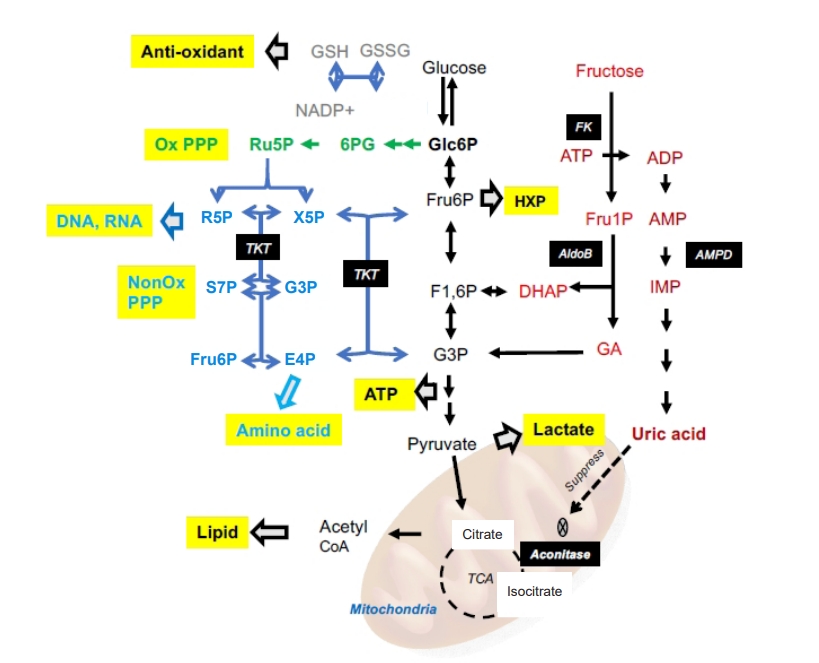
Several pathways downstream from fructose metabolism.
Fructose is initially metabolized to fructose 1-phosphate by fructokinase, which rapidly sequesters phosphate, consequently activating adenosine monophosphate (AMP) deaminase to cleave AMP to inosine monophosphate (IMP). Sequential enzymatic activation metabolizes IMP and eventually produces uric acid. Uric acid subsequently inhibits aconitase in the tricarboxylic acid (TCA) cycle to suppress mitochondrial respiration. Glycolysis is preferentially activated, and several metabolites are fed into several side pathways, including the pentose phosphate pathway (PPP) and hexosamine pathway (HXP), which aberrantly activate energy production, synthesis of biosynthetic precursors, and redox homeostasis.
ADP, adenosine diphosphate; AldoB, aldorase B; ATP, adenosine triphosphate; DHAP, dihydroxyacetone phosphate; E4P, erythrose 4-phosphate; F1,6P, fructose 1,6-biphosphate; FK, fructokinase; Fru1P, fructose 1-phosphate; Fru6P, fructose 6-phosphate; G3P, glyceraldehyde 3-phosphate; GA, glyceraraldehyde; Glc6P, glucose 6-phosphate; GSH, glutathione; GSSH, glutathione-S-S-glutathione; NADP+, nicotinamide adenine dinucleotide phosphate; NonOx, non-oxidative; Ox, oxidative; R5P, ribose 5-phosphate; Ru5P, ribulose 5-phosphate; S7P, sedoheptulose 7-phosphate; TKT, transketolase; X5P, xylulose 5-phosphate; 6PG, 6-phosphogluconate.
Glycolysis
The first enzyme involved in fructose metabolism is fructokinase (known as ketohexokinase [KHK]), which phosphorylates fructose to produce Fru1P (Fig. 4). There are two spliced isoforms of KHK, and each is produced by mutual exclusion of the adjacent exons 3C and 3A within the KHK gene [70]. The “A” isoform is ubiquitously expressed but has low activity due to relatively low affinity for its substrate (Km 8 mM) [71]. Expression of the “C” isoform is primarily restricted to metabolic tissues, including the liver, kidney, and intestine, and this form has much higher affinity for fructose (Km 0.8 mM) [71,72]. Fru1P is subsequently metabolized by aldolase B and triokinase to dihydroxyacetone phosphate and glyceraldehyde-3-phosphate that enter the glycolytic pathway downstream of phosphofructokinase. Subsequently, glyceraldehyde-3-phosphate is metabolized to pyruvate in the glycolytic pathway to produce ATP and nicotinamide adenine dinucleotide. Pyruvate is further converted into lactate by lactate dehydrogenase. Importantly, this reaction is usually stimulated by low oxygen, but is accelerated by fructose even under aerobic condition [73]. Lactate seems to be an energy for cancer growth [74].
Pentose phosphate pathway
The pentose phosphate pathway (PPP) is activated by fructose and comprises two distinct phases, the oxidative pathway and the non-oxidative pathway (Fig. 4). Glucose 6-phosphate, a fructose metabolite, is metabolized by three sequential reactions in the oxidative pathway to NAPDH, which supplies reducing equivalents, and reduces glutathione through the action of glutathione reductase. In turn, two forms of fructose carbon backbones, Fru6P and glyceraldehyde-3-phosphate, are catalyzed by transketolase to enter the non-oxidative pathway for nucleotide formation through ribose 5-phosphate, while erythrose 4-phosphate is metabolized into amino acids. Alternatively, activated hexokinase can convert fructose into Fru6P, which may be a link between glycolysis and the nonoxidative PPP in cancer cells [75].
Lipogenesis
Lipids are required as an energy source, for membrane formation, and as signaling molecules (Fig. 4). Fructose is metabolized in the glycolytic pathway to provide acetyl-CoA as the building block of carbon chains for de novo lipogenesis, and also promotes fatty acid synthesis to form palmitate. In turn, glyceraldehyde-3-phosphate, carrying a fructose-based carbon backbone, is also utilized to form triglycerides. Fructose also stimulates intracellular signaling pathways, including those mediated by carbohydrate-responsive element-binding protein [76] and GKRP [60]. A recent study using a mouse model demonstrated that fructose-mediated fatty liver disease was likely mediated by impairment of fatty acid oxidation due to an increased acetylation of long-chain specific acyl-CoA dehydrogenase and carnitine palmitoyl-transferase 1α [17].
Uric acid production and the Warburg effect
Fructokinase activation rapidly sequesters phosphate, consequently activating adenosine monophosphate (AMP) deaminase to cleave AMP to inosine monophosphate (IMP) (Fig. 4). However, phosphate levels subsequently increase due to the slower reaction of aldolase with Fru1P. This reaction is further accentuated by the increased IMP, which is an aldolase B inhibitor [77]. Sequential enzymatic activation metabolizes IMP and eventually produces uric acid. We found that uric acid could prevent the entry of fructose metabolites into mitochondrial oxidation in the human hepatocellular carcinoma cell line HepG2 [78]. A potential mechanism for this effect is the suppression of mitochondrial aconitase activity by uric acid, and disconnection of fructose metabolites from mitochondrial oxidation. Since aconitase lies at the junction of acetyl-CoA oxidation, blocking aconitase leads to acetyl-CoA shuttling out of the mitochondria, resulting in the accumulation of citrate in the cytosol. Citrate is then utilized for lipid synthesis by sequential ATP-citrate lyase and fatty-acid synthase [78]. As a result, fructose leads to a state of metabolic imbalance that favors glycolysis over mitochondrial respiration, resembling the Warburg effect in cancer [79].
The Warburg effect is shared by non-cancer disorders
In 1924, Otto Warburg initially described that cancer cells, as opposed to normal cells, exhibit a unique ability to ferment glucose to lactate even in the presence of sufficient oxygen [80]. This process is now recognized as a key mechanism of cancer growth and is called the “Warburg effect.” However, we need to be cautious for when interpreting this effect, as general scientists tend to share a misconception regarding oxidative metabolism in the mitochondria [81]. Warburg’s own experiments revealed persistent oxygen consumption in tumor tissues. The rate of mitochondrial respiration was low relative to what might have been predicted given the high rate of glucose uptake, but respiration itself did not appear to be impaired [81]. The Warburg effect was long considered a unique characteristic of cancer; however, recent studies indicate that aberrant glycolysis is not specific to cancer, but rather is shared by other non-cancer disorders [82].
The Warburg effect is involved in multiple processes in several disorders, and the cardiovascular, immune, and neuronal systems are now found to be all modulated by aerobic glycolysis [82]. Although glycolysis produces less ATP than mitochondrial oxidative phosphorylation, the process of aerobic glycolysis is much faster than that of mitochondrial respiration [83]. As a result, aerobic glycolysis can produce more ATP than mitochondrial oxidative phosphorylation in the same amount of time [84]. More importantly, the Warburg effect may impact more than energy production, and may regulate several cellular functions, including cell proliferation, extracellular matrix production, autophagy, and apoptosis [85], and may consequently participate in multiple biological processes.
The Warburg effect is involved in kidney diseases
Recent studies have documented that autosomal-dominant polycystic kidney disease (ADPKD) is mediated by aberrant glycolysis. Rowe et al. [86] demonstrated that cultured mouse embryonic fibroblasts derived from Pkd-/- mice exhibited activated glycolysis, given the cells preferentially utilized greater amounts of glucose and excreted more lactate into the culture medium than cells from wild-type mice. Mice lacking Pkd in the renal tubules, as a mouse model of ADPKD, exhibited glycolysis activation while blocking glycolysis with 2-deoxy-D-glucose (2DG), a glucose analog, attenuated tubular cell proliferation, leading to the reduction in kidney size and cyst formation [87].
In diabetic nephropathy, mitochondrial overproduction of superoxide due to the activation of the electron transport chain is considered a unifying mechanism, but this hypothesis remains controversial [88]. Recent studies have demonstrated that mitochondrial function is suppressed in diabetic nephropathy, and the restoration of normal mitochondrial health improves renal, cardiovascular, and neuronal outcomes. In addition, mitochondrial TCA cycle metabolites are also significantly reduced in patients with diabetic nephropathy compared to healthy controls [89]. In turn, glycolytic activation is upregulated in the renal cortex in type 2 diabetes [90], suggesting that activation of glycolysis is dominant over mitochondrial oxidation and plays a pathological role in diabetic nephropathy.
A shift to glycolysis has also been observed in two animal models; one is a model of unilateral ureteral obstruction and the other is a transforming growth factor (TGF)-β1-induced renal fibrosis model. Specifically, Ding et al. [91] found that myofibroblast activation in the kidneys was associated with enhanced renal glucose uptake and lactate production that could be attenuated by blocking glycolysis by 2DG treatment. In these models, a key factor is likely TGF-β1 as this growth factor was capable of switching metabolic profile favoring glycolysis over mitochondrial respiration in fibroblasts. In addition to TGF-β1, PDGF also causes the Warburg effect [92], consistent with the notion that growth factors disproportionately activate glycolysis relative to mitochondrial oxidation [93].
Could natural fruit exacerbate kidney disease?
One might ask whether fruit can be also deleterious to the kidney, given fruit contains substantial amount of fructose. This question arises from the assumption that fructose in natural fruit is theoretically metabolized, resulting in uric acid production, and therefore a large amount of fruit may be deleterious. In this regard, several clinical studies have examined the effect of fruit on renal function, and in many cases, the effect of consuming fruits together with vegetables, low salt, and other dietary modifications were assessed [94,95]. These studies generally found that our assumption was flawed and fruit was protective due to the improvement of metabolic acidosis, reduction of blood pressure, and prevention of cardiovascular diseases. However, the effect of a defined amount of fruits was examined in those studies, and it remains uncertain if a large amount of fruits could cause renal disease.
The mechanism by which fruits are protective of the kidney may be that metabolism of fructose in fruit is inhibited by vitamin C and other nutrients. For example, fructose-associated uric acid production is linked with xanthine oxidase activation and oxidant stress, which can be blocked by flavonoids/catechins and vitamin C in fruits [96]. Vitamin C also enhances urinary urate excretion through URAT-1 [97,98] and lessens the effects of uric acid. In addition, the potassium present in many fruits can ameliorate urate-induced endothelial dysfunction [99].
In this regard, we previously discussed this issue and reviewed the effect of variety of natural fruits on hyperuricemia or gout [100]. One thing to bear in mind is that the effects of fruit are often inconsistent in clinical studies, in part due to difference in study designs, although other factors may impact the results. Fruit intake is often estimated from the results of face-to-face interviews or questionnaires, which usually rely on memory, and may not always be accurate [101]. The composition of fruits may vary significantly depending on growing, harvesting, and storage conditions, and how they are prepared for consumption. For example, black currants become sweeter at higher growing temperatures and their taste varies with season, with the concentrations of fructose, glucose, and vitamin C found to also differ depending upon the season [102]. In addition, humidity and latitude influence the maturation process of many fruits, and often determine the sugar content; together, these various influences may result in sweeter fruit in the fall than in the spring, and greater fructose content in mature fruit than less mature fruit [103]. These factors may add a layer of complexity to the effects of fruit intake on human renal disease.
Conclusions
Several risk factors, including hypoxia, high blood glucose concentrations, senescence, and cardiac pressure overload, are found to share endogenous fructose production in the kidney as a common underlying factor (Fig. 5). A unique characteristic of fructose metabolism, as opposed to glucose metabolism, is the production of uric acid. Excessive uric acid production links to inflammation, endothelial dysfunction, vascular injury, and insulin resistance, while also favoring glycolysis over mitochondrial respiration, similar to the Warburg effect in cancer. The Warburg effect creates a pool of biosynthetic precursors that contribute to several pathological processes, including nucleotide synthesis, amino acid production, lipids and lactate, and excessive endogenous fructose is likewise a mechanism of CKD (Fig. 5). Further studies exploring the role of endogenous fructose in CKD are warranted.
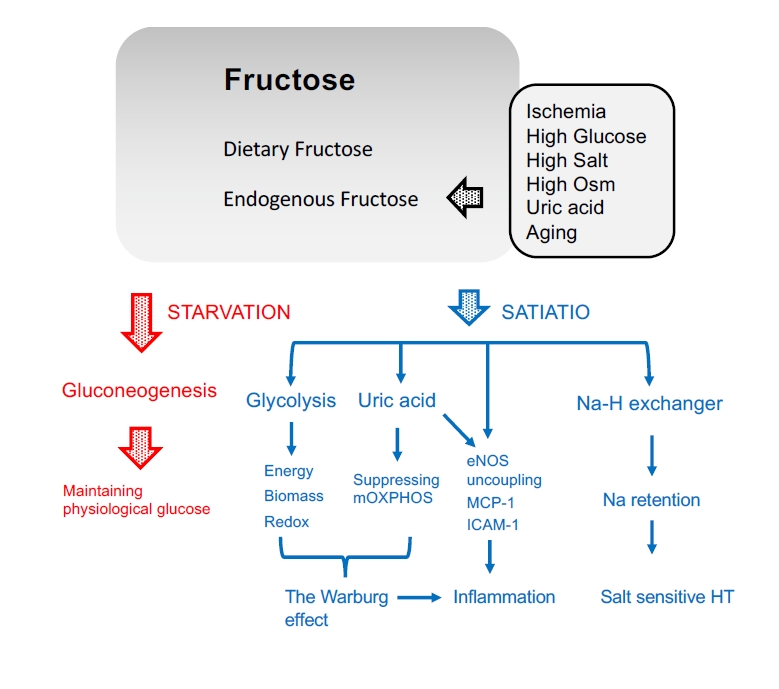
Current hypothesis of the pathophysiology of fructose in the kidney.
In addition to dietary fructose, fructose can be produced endogenously as a result of several pathological conditions in the kidney. During starvation, fructose is utilized for gluconeogenesis in the proximal tubular cells and excreted into the systemic circulation to maintain serum glucose concentrations. In turn, when in excess or during satiation, fructose is associated with aberrant energy production, biomass synthesis, and redox balance with uric acid production, resulting in the Warburg effect in the kidney. Together with such reactions, fructose also causes endothelial NO synthase (eNOS) uncoupling in endothelial cells and release inflammatory cytokines. Fructose also stimulates the Na/H exchanger to accelerate sodium absorption, leading to salt-sensitive hypertension (HT).
ICAM-1, intercellular adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1, mOXPHOS, mitochondrial oxidative phosphorylation; Osm, osmolarity.
Notes
Conflicts of interest
Takahiko Nakagawa has equity with XORTX therapeutics, which is developing novel xanthine oxidase inhibitors. The authors have no other conflicts of interest to declare.
Funding
This work was supported by a grant from the Korean Society of Nephrology (BAXTER, 2017) and a National Research Foundation of Korea (NRF) grant funded by the Korean government (MIST, (2020R1A2C3007759).
Authors’ contributions
Conceptualization: TN
Writing–original draft: TN
Writing–review & editing: DHK
All authors read and approved the final manuscript.