


Introduction
Autophagy is a highly conserved system by which damaged organelles, proteins, and lipids are removed by the cellŌĆÖs lysosomes. This essential system preserves homeostasis and cellular integrity in all eukaryotic organisms. Autophagy can be induced by various types of intra- and extracellular injuries, including those caused by inflammation, hypoxia, starvation, and endoplasmic-reticulum stress. Defective, excessive, and dysregulated autophagy are closely associated with the pathogenesis of metabolic disorder, inflammation, ischemia, aging, cancer, compromised immunity, and autoimmunity [1]. One study suggested that autophagy plays a protective role in renal disease [2]. The renoprotective role of autophagy has also been suggested in various animal studies, including those on acute kidney injury and aging [3ŌĆō5].
Autophagy involves the capture of cellular organelles and damaged proteins in autophagosomes, which ultimately lead them to lysosomes [6]. The generation of autophagosomes depends on the activation of numerous genes, including autophagy-related genes (Atgs), microtubule-associated protein 1 light chain 3 (LC3), and be-clin-1 genes [7]. Abnormal autophagy processes can alter renal tubule cells in diabetes mellitus (DM). The function of autophagy in DM nephropathy and high glucose (HG)-induced tubule injury remains mostly unknown. In addition, the changes that occur in intracellular autophagy in the HG milieu are unclear. Therefore, we evaluated the effects of HG on autophagy in human tubule cells.
Sulforaphane (1-isothiocyanoate-4-[methylsulfinyl]-butane, SFN) is an organic isothiocyanate [8]. It is a powerful activator of antioxidant proteins including heme oxygenase-1 (HO-1), NAD(P)H:quinine reductase, and glutathione transferase [9,10]. Antioxidant protein expression is controlled by the antioxidant response element (ARE) in its corresponding promoter region. Nuclear factor E2-related factor 2 (Nrf2) modulates the activation of AREs. Kelch-like ECH-associated protein 1 (Keap1) sequesters Nrf2 [11,12]. If oxidative stress modifies two crucial cysteine residues (C273 and C288) on the Keap1 protein, Nrf2 dissociates from the Nrf2/Keap1 complex. Dissociated Nrf2 migrates into the nucleus and attaches to the ARE sequences. This association between the Nrf2 and the ARE sequences activates the transcription of the phase II antioxidant proteins, including other Nrf2-responsive genes [12,13].
Reactive oxygen species (ROS) not only function as messengers in cell death, survival, and immunity pathways [14], but also can activate autophagy [15ŌĆō18]. SFN mediates antiproliferative and antioxidative processes (including cell death) caused by ROS generation in various cells [19ŌĆō21]. One group previously proposed that SFN-related ROS is an important factor in activating autophagy in pancreatic cancer [22].
We assessed the impacts of HG on the induction of autophagy using the HK-2 cells. We also evaluated the capability of SFN to preserve tubule cells from apoptosis by modulating autophagy. Our results demonstrate that SFN modulates autophagy through ROS-mediated extracellular signal-regulated kinase (phosphatidylinositol 3-kinase [PI3K]/Akt473 and p38 mitogen-activated protein kinase [MAPK]) activation in renal tubule cells.
Methods
Cell line and treatment
The HK-2 cell line was purchased from the American Type Culture Collection (Manassas, USA). The cells were processed with Gb3 combined with ╬▒-gal A (10 ╬╝g/mL), sphingolipid ceramide N-deacylase (SCDase, 10 ╬╝g/mL) or transforming growth factor (TGF)-╬▓ receptor inhibitor (SB 525334, 5 ╬╝M). None of the inhibitors was toxic at the doses used (assessed by cell viability assay). SFN was purchased from Calbiochem (San Diego, USA). SFN treatment was performed at 1.25 ╬╝m 1 hour prior to D-glucose stimulation. SFN was not toxic at the dose used (evaluated by cell viability assay, MTS assay; Promega, Madison, USA). The cells were simultaneously treated with different concentrations of D-glucose (5.5ŌĆō250 mM) for 1, 6, 12, 24, 48, and 72 hours.
Trypan blue assay and MTS assay
The cells were spread on 12-well plates at a density of 1.2 ├Ś 105 cells/cm2 in medium including either 5.5 or 250 mM glucose, assembled and stained with trypan blue. We calculated the full count of total cells and the number of trypan blue-stained cells. We prepared HK-2 cells in 96-well plates at a final volume of 100 ╬╝L/well culture medium for the MTS assay. The 10 ╬╝L/well Cell Proliferation Reagent WST-1 (Roche, Mannheim, Germany) was added. After incubating 1 to 4 hours at 37┬░C, we measured the absorbance of the samples at 420 to 480 nm.
Western blot assay
When the human HK-2 cells were grown to 60ŌĆō70% confluence, they were washed two times with phosphate-buffered saline (PBS) and lysed with lysis buffer (150 mM NaCl, 20 mM TrisŌĆōHCl, pH 7.5, 1% NP-40, 1 mM Na3VO4, 2.5 mM sodium pyrophosphate, 1 mM ethylene glycol tetraacetic acid, 1 mM NaF, 1 mM ethylenediaminetetraacetic acid and protease inhibitors cocktail; Sigma, St. Louis, USA) on ice for 0.5 hours. The washed cells were separated by 12% polyacrylamide gels and transferred to polyvinylidenedifluoride membranes. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour and visualized with an ECL detection system. They were exposed to Hyperfilm-MP (Amersham Pharmacia Biotech, Piscataway, USA) after 2 hours of incubation.
Immunofluorescence staining
The HK-2 cells were washed twice with cold PBS and fixed with 4% formaldehyde for 15 minutes. They then underwent permeabilization with 100% methanol. Following three extensive washings with PBS, the HK-2 cells were blocked with 1% normal fetal bovine serum in PBS buffer for 40 minutes at room temperature. They were subsequently incubated with anti-Lysotracker (In-vitrogen, Carlsbad, USA) and anti-LC3 (Cell Signaling Technology, Danvers, USA). They were later stained with Alexa fluor 488 goat anti-rabbit IgG (Life Technologies, Carlsbad, USA). Finally, the cells were double stained with DAPI to visualize the nuclei. All images were collected using a laser scanning confocal microscope (ZeissLSM510META; CarlZeiss, Oberko-chen, Germany).
Intracellular ROS detection
We measured the intracellular levels of ROS by flow cytometry using the ROS specific fluorescence probes 5-(and 6)-chloromethyl-2ŌĆÖ, 7ŌĆÖ-dicholrodihydrofluorescein diacetate, dihydroethidium (DHE; D11347) and acetyl ester (DCF; C6827) (Invitrogen). The tubule cells were incubated for 15 minutes at 37┬░C at a concentration of 5 ╬╝M DHE. Analyses were performed using a Cytomics 500 flow-cytometry with Cytomics RXP (Beckman Coulter, Miami, USA).
Statistical analysis
The data are reported as means ┬▒ standard deviation. The StudentŌĆÖs t test was used to compare groups. P values < 0.05 were considered statistically significant.
This study was conducted using an in vitro cell line. Neither human- nor animal-materials were used. Therefore, this study was waived the ethical approval and informed consents by the Institutional Review Board of Eulji University Hospital.
Results
Effect of HG on cell proliferation and apoptosis
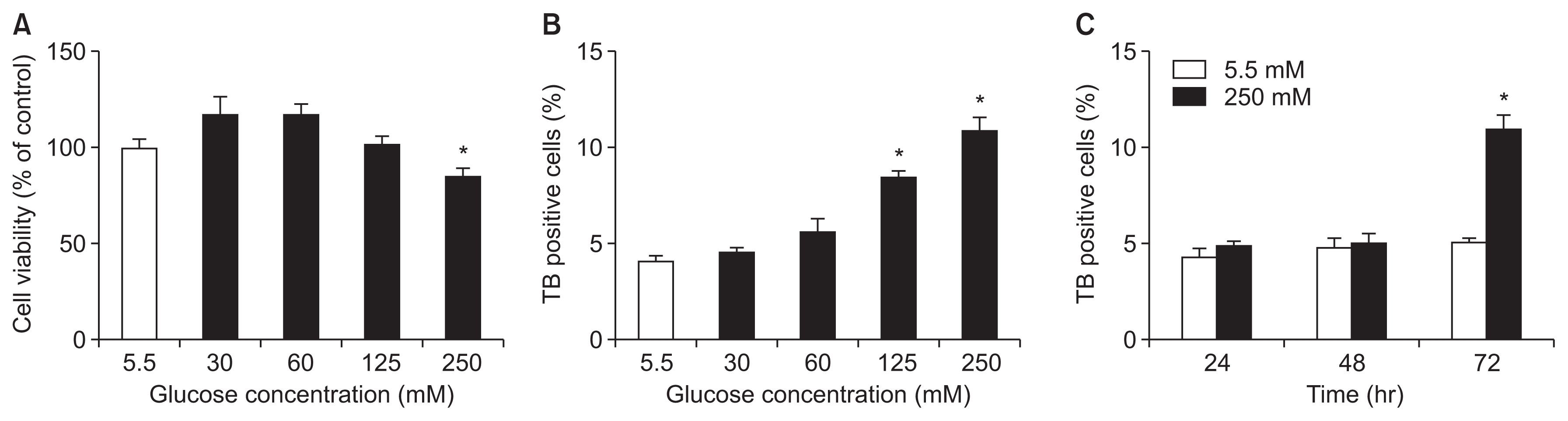
The MTS assay was used to evaluate cell viability and therefore examine the cytotoxicity induced by HG. Cell viability was significantly reduced in a glucose concentration-dependent manner (Fig. 1A, B). There was a significant decrease in viability after cells were cultured in 250 mM glucose compared to when they were cultured in 5.5 mM glucose (Fig. 1C).
The decrease in cell viability at the 250 mM concentration may have been caused by increased cell death or by reduced proliferation. When cell death was estimated, many dead cells were observed at 72 hours at the 250 mM concentration (Fig. 1C).
SFN modulates HG-induced autophagy and inhibits apoptosis
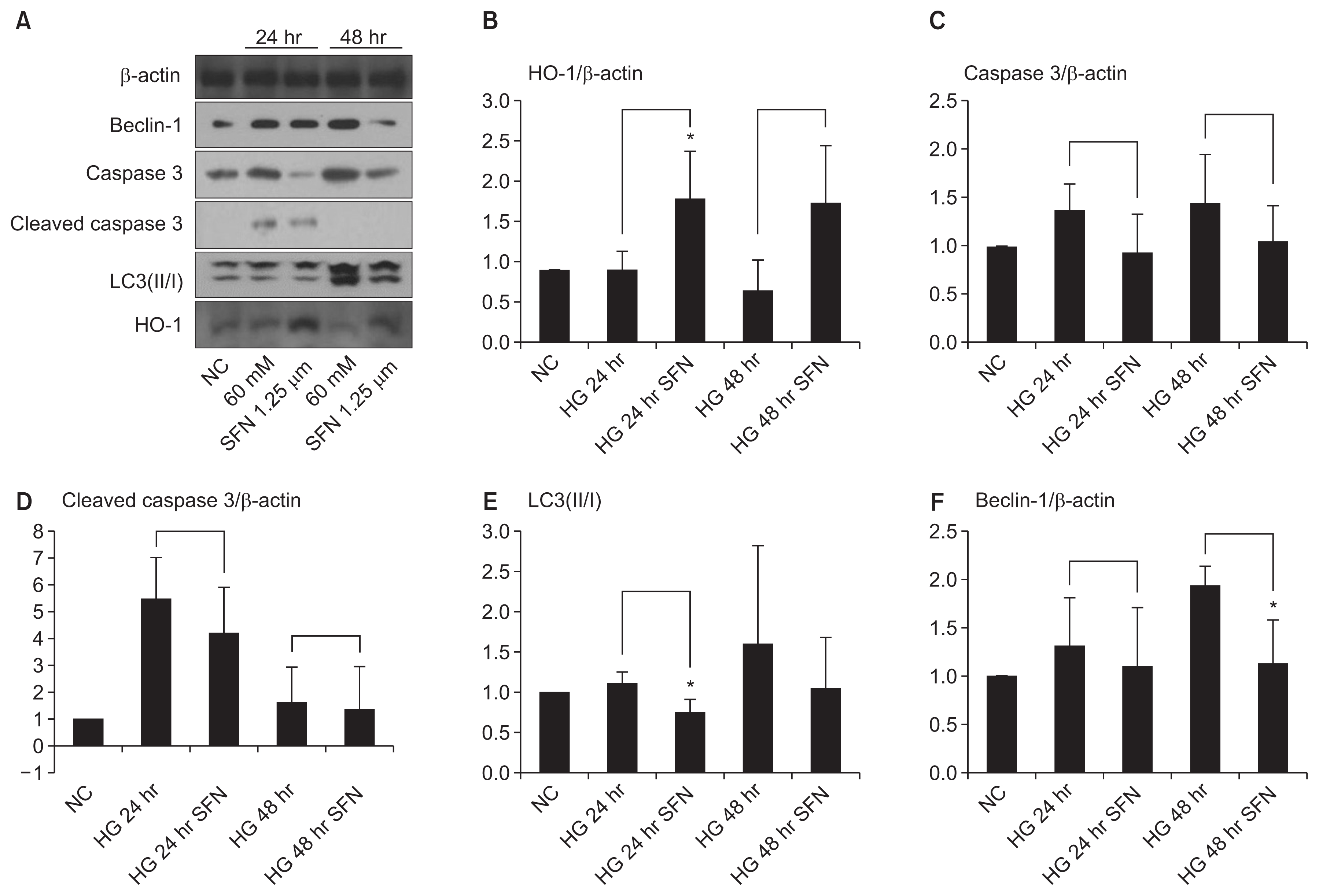
To study whether SFN influences autophagy and apoptosis in renal tubule cells, the HK-2 cells cultured in 60 mM glucose medium were treated with SFN (1.25 ╬╝M) for 1 or 2 days. The HG-stimulated cells showed increased expression of the autophagic markers beclin-1 and LC3(II/I) ratio (Fig. 2). HG also increased autophagy activity in HK-2 cells.
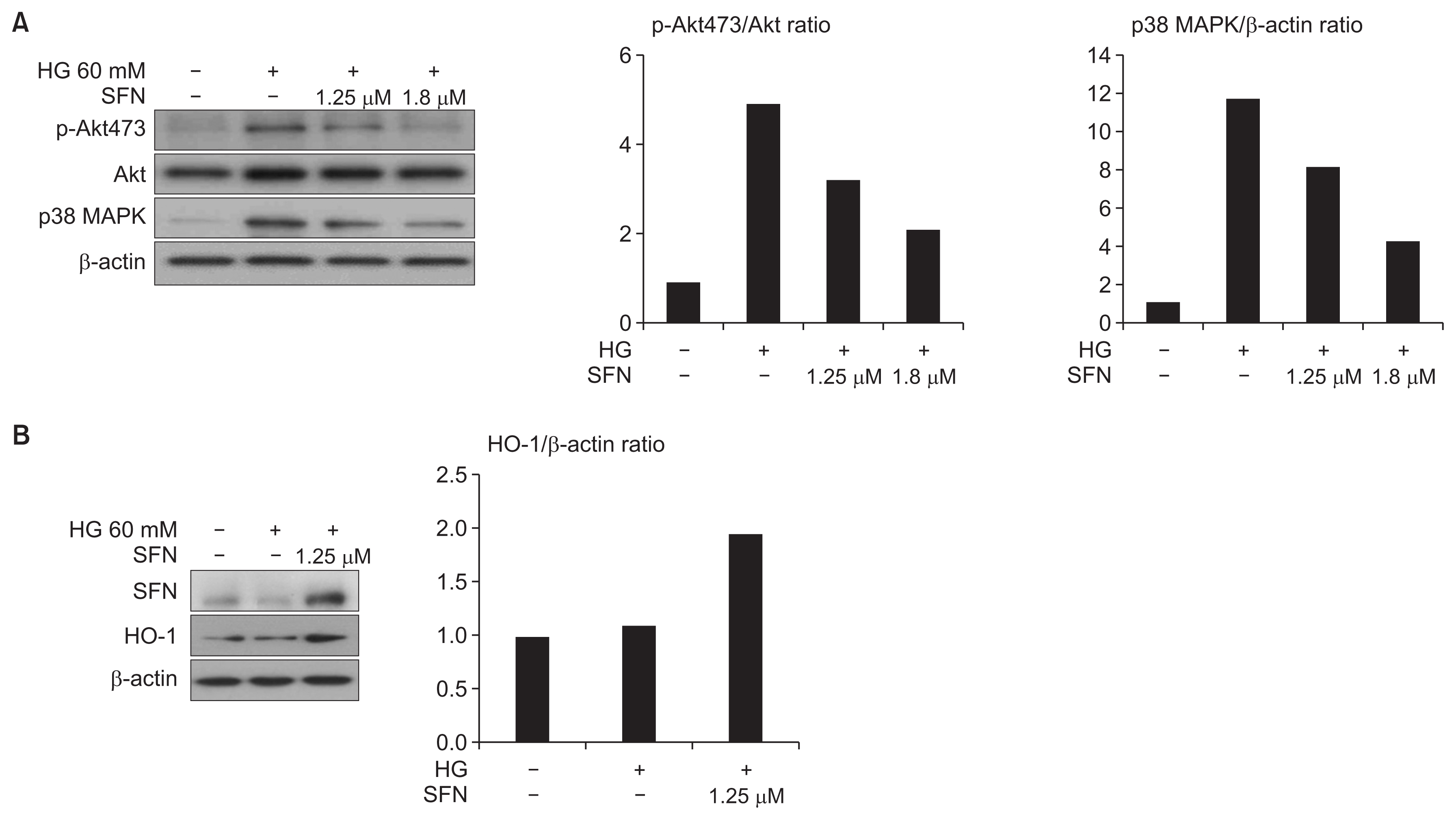
The expression of beclin-1 and the LC3(II/I) ratio in tubule cells treated with SFN was lower than that in the control group (Fig. 2). In addition, HO-1 was induced in HK-2 cells treated with SFN. There was also a reduction in the level of (activated) native and cleaved caspase-3 in the HG-induced cells, although the difference was not statistically significant (Fig. 2).
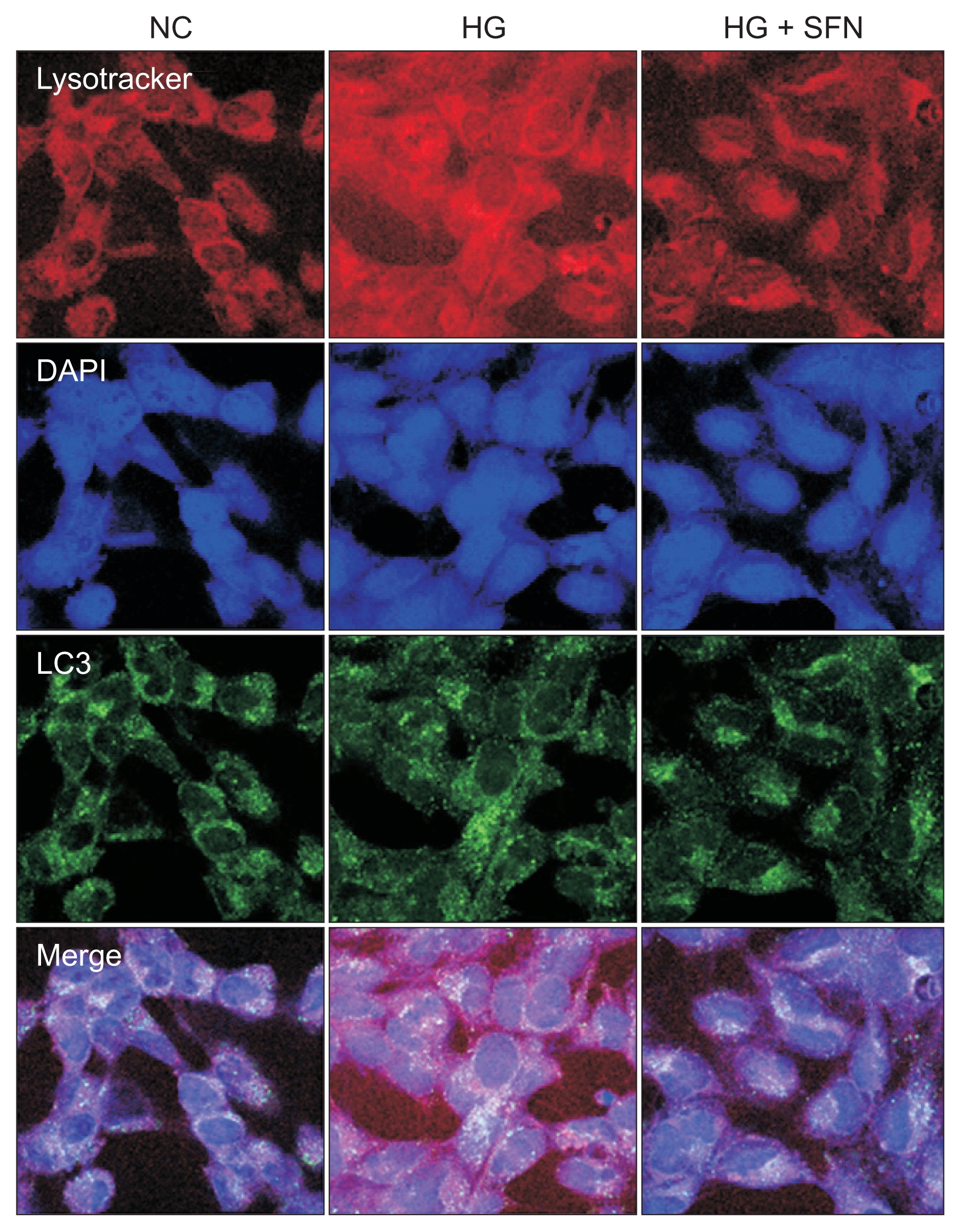
LysoTracker dyes were used to evaluate the autophagy process and to compare it with the activation of the microtubule-associated autophagy marker protein LC3. The immunofluorescence results were the same as those of the Western blot (Fig. 3).
These results collectively indicate that SFN modulates autophagy and may inhibit apoptosis.
ROS may mediate SFN-modulated autophagy
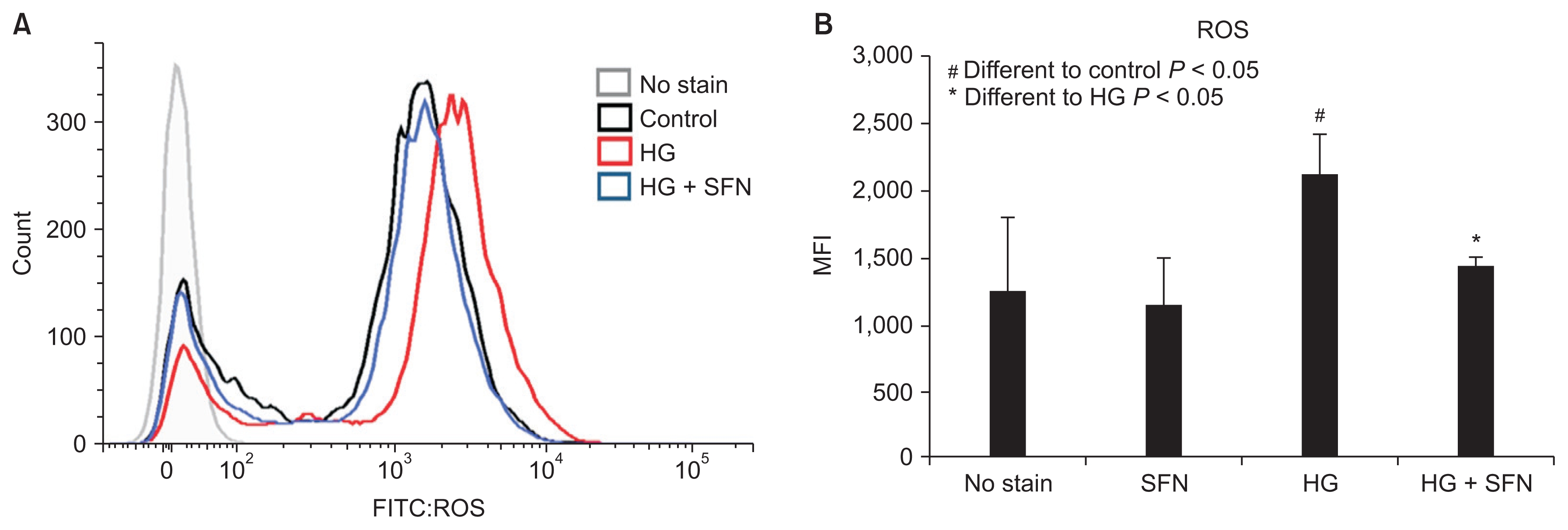
ROS are key inducers of autophagy and apoptosis. Therefore, we used FACScan (Beckman Coulter) to evaluate whether SFN could decrease the ROS levels in HK-2 cells. ROS levels were expectedly increased in HK-2 cells that were cultured in 60 mM glucose medium. In contrast, there was a decrease in the ROS levels in the cells that were treated with SFN (Fig. 4).
Involvement of the PI3K/Akt and p38 MAPK pathways in HG-induced autophagy and apoptosis
SFN treatment upregulated HO-1 expression and decreased the HG (60 mM)-induced activation of PI3K/ Akt473 and p38 MAPK (Fig. 5). We sought to confirm whether the HG-induced autophagy process involved the PI3K/Akt473 and p38 MAPK pathways. HG (60 mM) increased the expression of PI3K/Akt473 and p38 MAPK. These results support previous findings that have suggested that both pathways play important roles in HG-induced autophagy.
Discussion
The maintenance of the balance between tubule cell death and survival is essential to the development of DM nephropathy. We demonstrated HG-induced injury in renal tubule cells in vitro using several assays. Cell survival was considerably decreased, and cell death, including that by apoptosis, was markedly increased after exposure to HG.
The pro-autophagic effect of HG in this study involves the expression of beclin-1 and LC3. One previous study found that beclin-1 activation is involved with autophagy induction in cancer cells. The LC3, which is involved in autophagosome elongation, is the second essential protein. HG induced autophagy, as evidenced by the prompt accumulation of autophagic vacuoles in the cytoplasm and the increased expression of the autophagic marker LC3. The change in the autophagic response in renal tubule cells to HG-induced injury demonstrates that autophagy may be a therapeutic target for DM nephropathy. Autophagy was first regarded as a protective process under the condition of nutrient deprivation. However, several studies have suggested that autophagy plays a role in inappropriately enhancing cell death [23ŌĆō25], similar to the in vitro results observed in this study.
Activation of other antioxidant enzymes, including catalase and manganese superoxide dismutase, reduces autophagy and preserves cells from death in response to excess oxidative stress [26,27]. Cisplatin activates both HO-1 expression and autophagy. Activation of HO-1 decreases oxidative stress-induced cell death and prevents autophagy induction, while the lack of this protein increases ROS and apoptosis. Therefore, HO-1 modulates autophagy [28]. Another study found that HO-1 activation in lung epithelial cells decreased both ROS levels and autophagy during cigarette smoke extract-induced injury [29]. We similarly showed that HO-1 activation (in tubule cells) following SFN treatment was protective by considerably decreasing the HG-induced ROS generation, and thereby attenuating autophagy. This study suggests that the antioxidant effect of the Nrf2-HO-1 system may partially modulate autophagy. These data suggest that the Nrf2-HO-1 system preserves cells from apoptosis, both as a modulator of autophagy and as an antioxidant.
Several studies have demonstrated that autophagy can promote cell death or survival in response to the cellular environment or stress. One of the few disease processes in which autophagy inhibition may be beneficial is acute kidney injury due to sepsis. In a mouse model of lipopolysaccharide (LPS)-induced acute kidney injury, autophagy was induced in the renal cortex (that mainly consisted of tubules); in contrast, inhibition of autophagy protected the tubules against LPS-induced inflammation and acute kidney injury [30ŌĆō32]. However, the exact function of autophagy in renal disorders remains controversial, as studies have suggested that it is involved in both cell death and survival [33]. We believe that autophagy activation is advantageous in cells or tissues that require waste removal. In contrast, autophagy activation would adversely affect cells or tissues where physical barriers are important to protect against certain stimuli. Our results suggest that autophagy promotes tubule cell death and inhibits tubule cell growth in diabetic nephropathy with chronic HG injury. So far, it is clear that dysfunctional, overactive, or insufficient autophagy significantly contributes to renal injury.
The MAPK-activated signaling pathway was the first recognized to have the ability to mediate HO-1 activation caused by extracellular injury. The PI3K/Akt signaling pathway is also associated with HO-1 activation. The function of pro- and anti-inflammatory signaling cascades, such as p38 MAPK and PI3K/Akt, in HO-1 activation is very important [34]. We also demonstrated that both of these signaling pathways were involved in the Nrf2-HO-1 system.
Our findings demonstrate that activation of the Nrf2-HO-1 system can inhibit autophagy and prevent apoptosis. Therefore, the Nrf2-HO-1 system may be essential to maintain homeostasis through the modulation of autophagy and apoptosis.
In conclusion, activation of the Nrf2-HO-1 system inhibited ROS generation, concurrently attenuated autophagy, and reduced cell death after HG stimulation. HO-1 and autophagy are activated after exposure to an HG stimulus, and serve as a protective system. HO-1 activation inhibits ROS generation and oxidative stress during HG stimulation and considerably modulates autophagy and apoptosis. These results suggest that the Nrf2-HO-1 system may determine a cellŌĆÖs fate by decreasing oxidative stress and modulating autophagy during HG stimulation. Ultimately, the Nrf2-HO-1 system may be a novel therapeutic target in diabetic kidney disease.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









