


Introduction
Hepcidin was first isolated from human urine in the year 2000 and initially defined as solely an antimicrobial peptide [1], [2]. Although now accepted as the key regulator of iron balance, a role for hepcidin in iron metabolism was not recognized until 2001 when animal studies demonstrated that hepatic mRNA synthesis was induced by iron loading [3]. Since then, the structure, function, and regulation of hepcidin in normal iron balance, hereditary hemochromatosis, and in the anemias of chronic disease and chronic kidney disease (CKD) have been studied extensively. This review will discuss the physiology of iron absorption, the role of hepcidin in CKD-related anemia, and the possible diagnostic implications and limitations of using hepcidin as a marker.
The importance of iron and iron balance
Iron is the fourth most abundant element in the earth's crust, yet the average adult human body contains only 3ŌĆō4┬Āg, and iron deficiency is the most common cause of anemia worldwide. Since there are no significant physiologic mechanisms to regulate iron loss, iron homeostasis is dependent upon the tight link between intestinal iron absorption and total body iron requirements [4]. Intestinal iron absorption amounts to 1ŌĆō2┬Āmg of iron per day calculated from a dietary iron intake of 12ŌĆō18┬Āmg/day [5], and 1ŌĆō2┬Āmg of iron is lost on a daily basis through shedding of intestinal enterocytes, sweat, blood loss, and skin sloughing [6].
This intestinal iron absorption is essential as iron is a major component of heme in hemoglobin and myoglobin as well as an important cofactor in many redox enzymes [7]. Although crucial for life, free iron is toxic to cells [8], thus iron is maintained overwhelmingly in a bound or chelated state. Dietary iron absorption occurs almost exclusively in the duodenum, where it can be absorbed as heme (which is the most common dietary iron in Western cultures) or iron salts or nonheme (inorganic) irons such as ferrous sulfate [9].
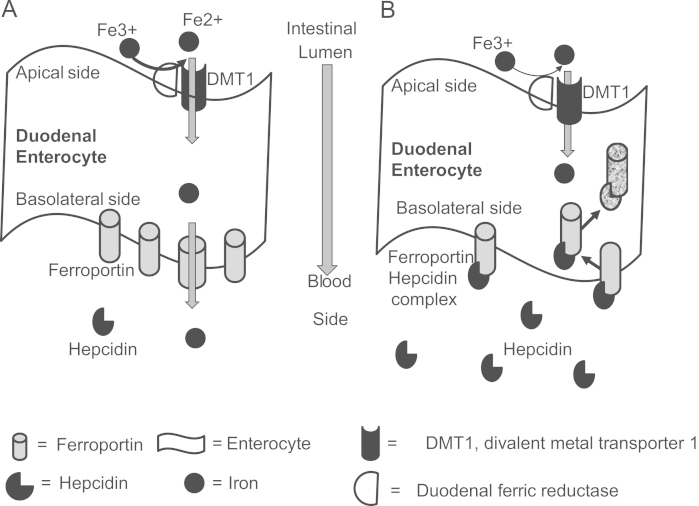
Heme iron is split from hemoglobin or myoglobin in the intestinal lumen and transported as intact iron porphyrin across the brush border [10]. Nonheme ferric iron (Fe3+) is reduced to ferrous iron (Fe2+) by a ferric reductase enzyme (also called duodenal cytochrome b) on the enterocytes' brush border [11], which then is transported across the apical enterocyte membrane by divalent metal transporter 1 (DMT1), a proton-coupled ion transporter, into the cell [5]. The possible fate of Fe2+ is to either be stored within intracellular ferritin or transported to the basolateral membrane, where it is exported by ferroportin1 into the plasma. Ferroportin is a transmembrane transporter for iron. Located in the basolateral membrane of duodenal cells and in the membrane of all iron exporting cells in the body, ferroportin exports Fe2+ from the cell.
Transport of iron from the cell to the plasma is facilitated by the oxidation of Fe2+ to Fe3+, which is catalyzed by the ceruloplasmin homolog hephestin [12]. Ferric iron is then rapidly bound to transferrin. This exquisite intestinal system provides iron to the body while protecting tissues against the oxidative effects of free iron. Co-ingested minerals may also alter intestinal iron absorption. Ascorbic acid can enhance nonheme absorption by reducing Fe3+ to the Fe2+ form, while calcium, zinc, and magnesium impair absorption. Iron deficiency anemia plays a large role in upregulation of duodenal DMT1, DcytB, and ferroportin1 mRNA levels along with protein expression [13].
The role of hepcidin and ferroportin in iron regulation
Hepcidin is a 25 amino acid peptide that was first identified in urine and plasma as an antimicrobial peptide [14]. It is synthesized in hepatocytes as a larger, inactive pre-prohepcidin, which is then cleaved by the prohormone convertase furin to generate mature hepcidin [15]. Hepcidin is encoded by the Hamp gene and its expression is promoted simultaneously by several factors, including transferrin-bound iron [16], pro-inflammatory cytokines [17], transferrin receptor 2 [18], hemojuvelin [19], the transcription factor Smad4 [20], HFE protein [21], and several bone morphogenic proteins [22].
The main role of hepcidin is to control the flux of iron into plasma by regulating ferroportin expression on the surface of cells. Under normal physiologic conditions, ferroportin is present on the basolateral membrane of duodenal enterocytes, and the surface of hepatocytes and macrophages. Surface expression of ferroportin is essential for iron to enter the systemic circulation (Fig. 1A). Hepcidin's main action is to bind to ferroportin, which results in internalization and degradation of the complex, effectively preventing duodenal iron absorption and reducing iron release from macrophages (Fig. 1B) [23]. Because duodenal cells are eventually sloughed, intestinal iron absorption is prevented, while decreased release of iron from storage results in reduced circulating plasma iron.
Hepcidin levels decline when iron stores are low, permitting enhanced iron absorption. Hepcidin levels increase in response to high iron stores and high serum iron, and thus protects against iron overload [24], [25]. This was demonstrated in a mouse model in which constitutive overexpression of hepcidin resulted in death from iron deficiency anemia [26]. A serine protease also expressed in the liver, TMPRSS6, is thought to participate in a transmembrane signaling pathway that is triggered by iron deficiency anemia to block transcription of the gene for hepcidin, Hamp, resulting in lower hepcidin production in order to permit dietary iron absorption [27].
Aside from iron, hepcidin is also clinically regulated by anemia, hypoxia, and inflammation [14]. Human hepatocytes increase hepcidin mRNA in the presence of IL-6 or lipopolysaccharide and in the presence of IL-6 produced by monocytes exposed to lipopolysaccharide [17]. In one human case study, infection increased the excretion of urine hepcidin 100-fold [17]. As an acute phase reactant, hepcidin activity therefore mirrors ferritin, which makes the interpretation of iron studies in the presence of inflammation very difficult [7], [28]. Lastly, hypoxemia and anemia appear to suppress hepcidin via the stimulation of epoetin, which improves iron mobilization as compensation [29].
Hepcidin in the CKD and dialysis populations
Given the confounding variables in managing and evaluating anemia of CKD [28], the nephrology community has long been searching for a novel marker and predictor for iron responsiveness. Our current biomarkers of ferritin and transferrin saturation (TSAT) are often subpar, and hepcidin has thus been postulated to offer more definitive diagnostic promise as it is the master regulator for iron absorption and release of iron from reticuloendothelial stores (RES).
Many studies have demonstrated that hepcidin levels increase progressively with severity of CKD, with predialysis CKD patients having a two- to four-fold elevation of hepcidin, and dialysis patients with a six- to nine-fold increase of hepcidin [30]. Some studies have failed to demonstrate elevated hepcidin in early CKD, and this could reflect differing populations, the hepcidin assays employed, and the power of the study to detect differences given the wide intrapatient short-term variability in hepcidin levels [31].
Several investigators have found that hepcidin and ferritin correlate strongly in dialysis patients [32], and it has previously been hypothesized that the inverse relationship of glomerular filtration rate (GFR) and hepcidin is due to the known association of CKD and inflammation. Although ferritin, an acute phase reactant, is a marker of iron stores and inflammation, it has continually lacked a strong predictive value for identifying iron responsiveness [33].
The correlation between markers of inflammation and hepcidin levels has been demonstrated in several studies that may actually preclude its clinical utility for iron status evaluation in patients with inflammation [31], [34]. This was also supported by a study by Zaritsky and colleagues in 2009, when the authors included erythrocyte sedimentation rate and high sensitivity-C reactive protein (CRP) covariates in their multivariate analysis and concluded that the relationship between inflammation and hepcidin levels persisted [35]. Not all studies, however, support this correlation between hepcidin and inflammatory markers [36], [37].
Furthermore, some studies have suggested no difference in hepcidin levels between epoetin-responsive and nonresponsive dialysis patients and no correlation between hepcidin and epoetin dose [31], [36]. While some have reported an inverse relationship between hepcidin and epoetin with a corresponding decline in hepcidin levels when epoetin is initiated, the variability in hepcidin levels suggests the relationship is insufficient to guide clinical decisions regarding iron and erythropoiesis-stimulating agent (ESA) dosing [37], [38].
Animal models have suggested that elevated hepcidin levels impair response to even large ESA doses [39], raising suspicion that high hepcidin levels contribute to the ESA resistance observed in some chronic hemodialysis patients. This raises the possibility that lowering hepcidin levels or impairing hepcidin interaction with ferroportin could be an effective anemia treatment in CKD and dialysis patients.
IV and oral iron: overcoming hepcidin-mediated blockade
Elevated hepcidin contributes to the anemia of CKD and the anemia of chronic inflammation by binding to the ferroportin channel and inhibiting the release of iron from macrophages, hepatocytes, and enterocytes. In time, this chronic elevation of hepcidin may result in iron deficiency, but even in the short term, hepcidin blockade results in ŌĆ£functionalŌĆØ iron deficiency due to impaired release of iron from the RES. Due to this, even patients with adequate iron stores can experience iron-restricted erythropoiesis, especially when red blood cell production is increased by ESAs [40].
Intravenous (IV) iron supplementation has been found to overcome hepcidin-mediated blockade and iron-restricted erythropoiesis by improving anemia, including patients with elevated levels of hepcidin as well as elevated CRP levels [41], [42]. IV iron can be released immediately by the macrophages and saturate plasma transferrin or become stored in ferritin [43]. This effect, however, was not seen with oral iron supplementation, presumably because hepcidin has internalized the basolateral ferroportin on duodenal cells, thereby preventing absorption.
Suppressing hepcidin's actions could reestablish the efficacy of oral iron therapy for treatment of iron-restricted erythropoiesis, and enhance release of any iron present in the RES. Administration of an antihepcidin antibody has been found in an animal model expressing human hepcidin to treat inflammation-induced anemia [39]. Also, using a small-interfering RNA has been shown to directly suppress hepcidin transcription and increase serum iron levels [39]. Several potential targets involved in hepcidin signaling and transcription, such as anti-bone morphogenic protein (anti-BMP) molecules, STAT3 inhibition, and HIF promoters, also can reduce hepcidin expression [44], [45], [46].
The average hemodialysis patient loses 1.5ŌĆō3.0┬Āg of iron per year, or 30ŌĆō60┬Āmg of iron per week. Clinical trials to date have not demonstrated long-term harm from IV iron therapy in dialysis patients, but trials have not been ideally designed to fully assess safety. An IV iron supplementation strategy will increase hepcidin levels further, and could adversely affect long-term iron mobilization. As hemodialysis patients have continual blood (and therefore iron) losses, the goal of IV iron treatment should be to first replete the iron stores, then provide sufficient IV iron to match ongoing losses.
Most dialysis patients receiving IV iron have also been found to have hemosiderosis as determined by magnetic resonance imaging (MRI) [47]. However, these studies are controversial because such imaging cannot differentiate between IV iron taken up into the RES by the liver and the pathological deposition of iron into liver parenchymal cells.
There are several lines of evidence to suggest IV iron in hemodialysis patients is not leading to iron overload-mediated organ damage. There is a relatively rapid dissipation of liver iron content as measured by MRI scanning in hemodialysis patients, suggesting either surprisingly large ongoing blood losses, which is highly unlikely, or the relationship of total body iron content to MRI results is fundamentally different in hemodialysis patients receiving IV iron products compared with patients with transfusional iron overload. Patients with transfusional iron overload and end-organ damage have 15ŌĆō25┬Āg of excess body iron, which is more than the average hemodialysis patient receives in a lifetime, even before accounting for the substantial iron losses due to ongoing blood loss. Despite the marked increase in IV iron use and serum ferritin levels in the United States and Europe, there have not been reports of liver or heart damage related to iron overload. Nevertheless, all therapies have risks, and prudent dosing of IV iron should reflect an assessment of estimated iron needs and ongoing iron losses. Lastly, aggressive treatment of anemia with ESA does not reduce cardiovascular events or lower mortality, and therefore the urgency to treat anemia is largely misplaced.
Practical clinical implications of hepcidin testing
Several serum hepcidin assays have been developed that correlate well to iron stores in select populations, while urinary hepcidin and serum prohepcidin have not been clinically useful [48], [49]. Furthermore, there is a wide variability (as much as a 10-fold difference) in the level of hepcidin values reported by various methods including surface enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS), liquid chromatography mass spectrometry, competitive enzyme-linked immunosorbent assay (ELISA), and competitive radioimmunoassay (RIA) [50]. The reasons for these discrepancies are unclear, but possible etiologies include cross-reactivity with hepcidin metabolites, hepcidin-binding factors in the serum, and different hepcidin standards.
In clinical practice, a low transferrin saturation (<25%) coupled with low ferritin (<200┬Āng/mL in dialysis patients, and <100┬Āng/mL in CKD Stage 3 and 4 patients) indicates a high likelihood of iron deficiency, especially if the patient is anemic and receiving an ESA. Such patients usually have low hepcidin levels. Due to the direct relationship between ferritin and hepcidin, and the broad short-term intrapatient variability in hepcidin, it is unlikely hepcidin offers any better predictive value than ferritin. Several investigations to date support this view [31], [34], [51]. Similarly, a hepcidin determination is not required in iron-replete patients exhibiting high transferrin saturation and high ferritin.
The most difficult anemic patients to manage have low transferrin saturation coupled with high ferritin, suggesting the anemia is due to iron deficiency, inflammation, or a combination of those factors. Unfortunately, such patients also have high hepcidin levels, and therefore, hepcidin does not discriminate among these disorders and cannot guide therapy better than our standard testing. Despite being correlated with ferritin, hepcidin has been found to be no better than ferritin in predicting iron stores or iron requirements in hemodialysis patients. Until the contribution of inflammation to high hepcidin levels is eliminated or quantified, hepcidin offers no superiority over ferritin and transferrin saturation as markers of iron stores or iron needs.
Conclusion
Hepcidin has been found to be the key regulator of iron homeostasis and plays a key role in CKD and chronic disease anemia as it is excreted by the kidneys and regulated by inflammation. At present, the measurement of hepcidin is erratic and does not provide any current diagnostic value over ferritin and other available iron studies. This target continues to be widely studied and may hold promise as an important biomarker of iron metabolism and a therapeutic target in the future.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









