Proximal renal tubular acidosis with and without Fanconi syndrome
Article information
Abstract
Proximal renal tubular acidosis (RTA) is caused by a defect in bicarbonate (HCO3−) reabsorption in the kidney proximal convoluted tubule. It usually manifests as normal anion-gap metabolic acidosis due to HCO3− wastage. In a normal kidney, the thick ascending limb of Henle’s loop and more distal nephron segments reclaim all of the HCO3− not absorbed by the proximal tubule. Bicarbonate wastage seen in type II RTA indicates that the proximal tubular defect is severe enough to overwhelm the capacity for HCO3− reabsorption beyond the proximal tubule. Proximal RTA can occur as an isolated syndrome or with other impairments in proximal tubular functions under the spectrum of Fanconi syndrome. Fanconi syndrome, which is characterized by a defect in proximal tubular reabsorption of glucose, amino acids, uric acid, phosphate, and HCO3−, can occur due to inherited or acquired causes. Primary inherited Fanconi syndrome is caused by a mutation in the sodium-phosphate cotransporter (NaPi-II) in the proximal tubule. Recent studies have identified new causes of Fanconi syndrome due to mutations in the EHHADH and the HNF4A genes. Fanconi syndrome can also be one of many manifestations of various inherited systemic diseases, such as cystinosis. Many of the acquired causes of Fanconi syndrome with or without proximal RTA are drug-induced, with the list of causative agents increasing as newer drugs are introduced for clinical use, mainly in the oncology field.
Introduction
Proximal renal tubular acidosis (RTA) as an isolated defect in tubular transport of bicarbonate (HCO3−) is characterized by a decreased rate of HCO3− reabsorption in the proximal tubule in the absence of alterations in the transport of other solutes [1–8]. Unlike in patients with classic distal RTA, patients with proximal RTA are able to lower their urine pH when plasma bicarbonate falls to a certain level [4,8]. Isolated proximal RTA is exceedingly rare, with the exception of the iatrogenic form, which is caused by the use of carbonic anhydrase (CA) inhibitors. Proximal RTA is more commonly associated with generalized dysfunction of the proximal tubule as part of Fanconi syndrome [2–4]. The syndrome is caused by a generalized dysfunction of the reabsorptive capacity of the kidney proximal tubule, causing a loss of solutes, including phosphate, uric acid, glucose, amino acids, low-molecular-weight proteins, and bicarbonate [9–12].
In recent years, new causes of drug-induced RTA and Fanconi syndrome have been recognized and the mechanisms of the proximal tubular alterations causing Fanconi syndrome are much better understood. In this review, we discuss proximal RTA, including the many causes of drug-induced proximal RTA and Fanconi syndrome.
Overview of bicarbonate transport along the nephron
In an individual with normal plasma HCO3− level and a glomerular filtration rate (GFR) of 100 mL/min, ~2,500 mEq of HCO3− is filtered daily; virtually all of this HCO3− is reabsorbed so that essentially none appears in the urine [2,13]. Bicarbonate is freely filtered by the kidney glomerulus, and its concentration in the glomerular filtrate is equal to that in plasma (~25 mEq/L) [2,13]. The majority (~80%) of the filtered HCO3− is reabsorbed in the proximal tubule and the remaining 20% that escapes reabsorption is reclaimed distally by the thick ascending limb (TAL) of the loop of Henle, distal tubules, and collecting tubules. Because the vast majority of HCO3− is normally reabsorbed in the proximal tubule, finding a significant amount of HCO3− in the urine is usually taken as evidence of a defect in proximal tubular HCO3− reabsorption. When there is a major defect in HCO3− reabsorption in the proximal tubule, a larger quantity of filtered HCO3− is delivered to the distal segments, including the TAL, overwhelming the distal system and causing urinary HCO3− wastage, which is the clinical hallmark of underlying derangement in proximal tubular function.
The mechanisms involved in HCO3− reabsorption in proximal tubular cells are depicted in Fig. 1. HCO3− transport across the luminal membrane does not take place as such; rather it is the result of secretion of H+ into the tubular lumen. The filtered HCO3− reacts with the luminal H+, which is secreted mainly by an apical sodium/hydrogen exchanger 3 (Na+/H+ exchanger isoform 3 [NHE3]) (solute carrier 9 A3 [SLC9A3]) [14] and to a lesser extent by an apical vacuolar H+-ATPase [15] to form H2CO3. This weak acid is readily dissociated into CO2 and H2O by a brush border CA. The kidney has at least two forms of carbonic anhydrase, CA II and CA IV [16]. CA II is found in the cell cytoplasm of both proximal and distal tubules and is also present in red blood cells, whereas CA IV is found mainly in the brush border of the proximal tubule. The latter is the isoform of CA involved in facilitating apical HCO3− reabsorption in the proximal tubule. It performs this function by preventing the luminal accumulation of H2CO3, creating a more favorable pH gradient for H+ secretion (Fig. 1) [4,5].

Bicarbonate reabsorption mechanisms in the proximal tubular cell.
On the luminal side, the Na+-H+ exchanger, NHE3, carries out the majority (~80%) of hydrogen ion secretion, while the proton pump H+-ATPase is responsible for ~20% of proton transport. Note that CO2 entry via an AQP1 channel is controversial and not fully proven.
AQP1, aquaporin 1; CA, carbonic anhydrase; NBCe1-A, sodium bicarbonate cotransporter 1 variant A; NHE3, Na+/H+ exchanger isoform 3. Modified from Singh et al [4] with permission.
The luminal production of CO2 from the dehydration of H2CO3 is accelerated by glycosylphosphatidylinositol (GPI)-anchored CA IV enzyme [17]. The resulting water molecule (H2O) passes through the apical aquaporin channel. While it has long been assumed that CO2 diffuses freely across the cell membrane, recent evidence suggests a role for the apically expressed aquaporin 1 (AQP1) in the proximal tubule in facilitating the entry of CO2 to proximal tubular cells [18,19]. However, experiments by Fang et al [20] provide evidence against AQP1–dependent CO2 permeability in the lung and kidney. More studies are needed to define the role of aquaporin channels in the entry of CO2 to cells.
Following the flux of CO2 into the proximal tubular cells, a reverse hydration reaction catalyzed by the cytoplasmic CA II yields H+ and bicarbonate ions [13,16,21]. While the formed H+ is recycled to the apical side, HCO3− is extruded with Na+ across the basolateral cell membrane via the electrogenic sodium-bicarbonate cotransporter (NBCe1-A [SLC4A4]) [6,22,23]. The driving force for this HCO3− and Na+ efflux is the basolateral membrane potential, which is generated by Na+/K+ ATPase and the twin-pore domain acid-sensing K+ 2 (TASK2) K+ channels [23,24].
Bicarbonate escaping reabsorption by the proximal tubule [25,26] is later reclaimed by the TAL via a similar process involving NHE3. Approximately 15% of the filtered bicarbonate load is reabsorbed by the TAL [27]. Apical NHE3 in the TAL functions similarly to its proximal tubule counterpart and is the main contributor to HCO3− reabsorption in this segment of the nephron. A small but significant contribution of apical vacuolar H+-ATPase also occurs here [27]. Once HCO3− is regenerated intracellularly from H2CO3 (catalyzed by CA II and CA XV [6]), it exits through the basolateral membrane of the TAL cell mainly via the Cl−/HCO3− exchanger (AE2 anion exchanger) [6,27] and the K+/HCO3− cotransporter [27].
In the distal nephron and collecting tubules, acid secretion and thus bicarbonate reabsorption is achieved by the α-intercalated cell, whereas bicarbonate secretion occurs in the β-intercalated cell [8]. In α-intercalated cells, urinary acidification involves a combination of energy-dependent proton secretion across the apical surface mediated mainly by a hydrogen ATPase and, to a lesser extent, hydrogen potassium ATPase [8]. Basolateral chloride-bicarbonate exchange serves to transport bicarbonate back into the blood via a band-3 protein, AE1, which is also critically important for continued acid excretion via the apical H+-ATPase and H+/K+-ATPase pumps [28–31].
Isolated proximal RTA
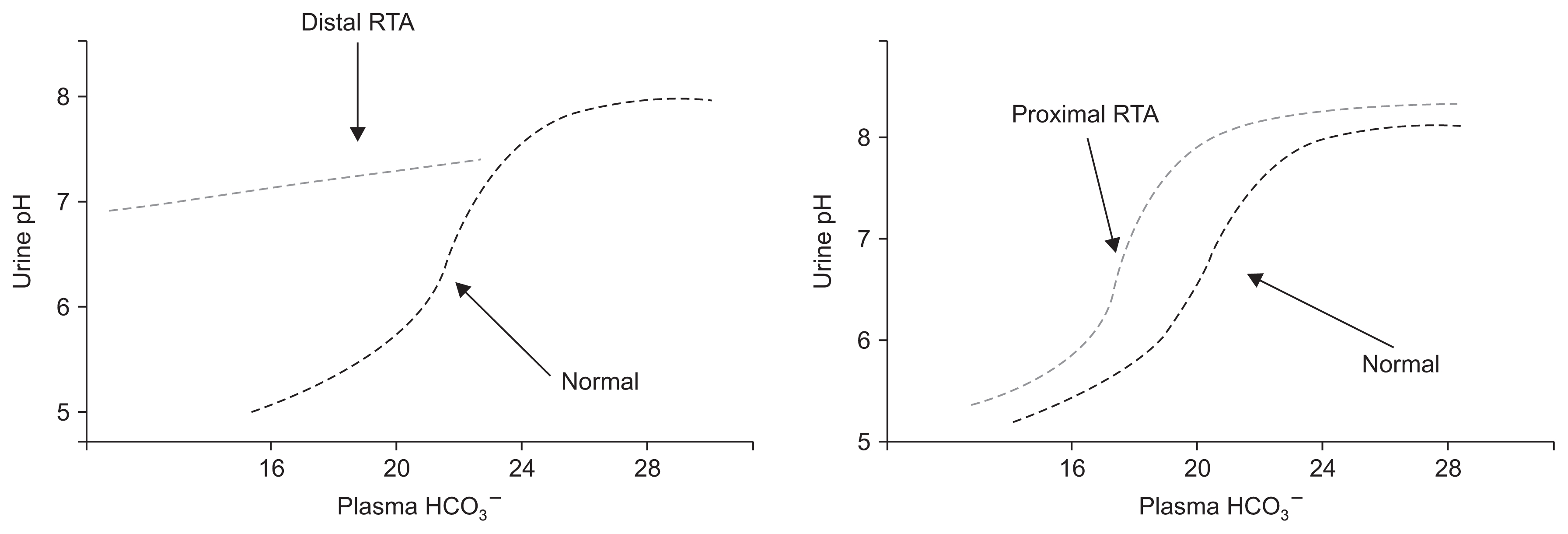
Impaired HCO3− reabsorption in proximal RTA can be understood as a decrease in the renal threshold for HCO3− reabsorption[1,2,32]. In this form of RTA, proximal HCO3− reabsorption can only accommodate a portion of the filtered HCO3−. The portion “rejected” by the proximal tubule cannot be totally reclaimed distally and therefore appears in the urine in large amounts when plasma HCO3− is above a given threshold (usually 16–20 mEq/L). If plasma HCO3− falls below this level, there is sufficient capacity for proximal tubular HCO3− reabsorption such that urine HCO3−, and thus urine pH, can fall to near normal levels (Fig. 2) [1,2,32]. Isolated proximal RTA can be hereditary or acquired as discussed below.
Hereditary proximal RTA
Isolated proximal RTA can be autosomal dominant, autosomal recessive, and sporadic. The autosomal recessive type is associated with severe growth retardation, ocular abnormalities such as glaucoma, cataracts and band keratopathy, and mental retardation [23,33]. This autosomal recessive type has been associated with mutations in the basolateral NBCe1 [23,33]. Mutations in this transporter lead to reduced activity and/or trafficking, disrupting the normal bicarbonate reabsorption process of the proximal tubules.
To our knowledge, autosomal dominant proximal RTA has been reported only in a single Costa Rican family [2,34]. The clinical features include mild growth retardation and reduced bone density. Sporadic isolated proximal RTA is a non-familial transient disorder that has been reported during infancy [2,35]. Patients with this disorder experience defective renal and intestinal bicarbonate reabsorption [33].
An isolated defect of proximal tubular HCO3− reabsorption is also caused by the use of CA inhibitors. A deficiency or a malfunction in luminal CA IV could lead to impairment of proximal HCO3− reabsorption but, to our knowledge, CA IV deficiency has not been demonstrated in patients with hereditary proximal RTA. By contrast, familial RTA has been long recognized in patients with inherited CA II deficiency in red blood cells [5,36–38]. These patients had features of both proximal and distal RTA (type III RTA) as well as osteopetrosis, cerebral calcification, and mental retardation.
Hereditary distal RTA with associated features of proximal tubular dysfunction (low-molecular-weight proteinuria, generalized hyperaminoaciduria, hypophosphatemia with hyperphospaturia, and hypouricemia with hyperuricosuria) was described in two siblings with ATP6V1B1 mutation [38]. This rare association is not fully understood, although it may go away after correction of the hypokalemia.
Acquired proximal RTA
CA inhibitors, such as acetazolamide are often used to manage conditions such as glaucoma (by reducing intraocular pressure), mountain sickness, or increased intracranial pressure [39] and are a well-recognized cause of isolated proximal RTA [9]. Three CA inhibitors (acetazolamide, methazolamide, and dichlorphenamide) can be administered systemically. The other two CA inhibitors (brinzolamide and dorzolamide) are applied topically. All CA inhibitors are sulfonamide derivatives with the potential to cause proximal RTA [39]. The defect in HCO3− reabsorption with CA inhibitors can be explained by inhibition of CA IV located in the apical membrane of the proximal tubule cells. Some CA inhibitors, such as acetazolamide and benzolamide, are less membrane-permeable than others and as such are not as effective inhibitors of cytosolic CA as membrane-bound CA [40]. CA inhibitors typically cause pure proximal RTA as a result of inhibition of the membrane-bound CA IV isoform, resulting in the isolated inhibition of HCO3− reabsorption without any associated features of Fanconi syndrome [41].
Fanconi syndrome
The inherited causes of Fanconi syndrome can be primary or secondary to systemic diseases (Table 1). Most often, however, Fanconi syndrome is the consequence of drug-induced nephrotoxicity.

Inherited causes of Fanconi syndrome
Primary Fanconi syndrome
Primary Fanconi syndrome is usually caused by a missense mutation in the sodium phosphate cotransporter (NaPi-II) in the proximal tubular apical membrane [42]. A new form of inherited Fanconi syndrome recently described by Klootwijk et al [43] in an extended black family is caused by a mutation of the EHHADH gene, an enzyme involved in peroxisomal oxidation of fatty acids and expressed in the proximal tubule. This mutation leads to impaired oxidative phosphorylation and a reduction in ATP available in proximal tubular epithelial cells, which results in turn in defects in the transport of molecules across the proximal convoluted tubule (PCT) cells [43].
Another recently reported cause of Fanconi syndrome is a mutation in the HNF4A gene [44]. HNF4A encodes a member of the nuclear receptor superfamily of ligand-dependent transcription factors. Most of the information on this gene come from studies in hepatocytes, but its role in the kidney is not completely understood. The R76W mutation in HNF4A causes additional features not seen in Fanconi syndrome: nephrocalcinosis, renal impairment, hypercalciuria with relative hypocalcemia, hypermagnesemia, neonatal hyperinsulinism, and macrosomia [44].
Fanconi syndrome associated with inherited systemic disease
The secondary causes of Fanconi syndrome include inherited cystinosis, galactosemia, hereditary fructose intolerance, tyrosinemia, Lowe syndrome, Alport syndrome, Wilson disease, and mitochondrial disorders (Table 2). The most common inherited cause of Fanconi syndrome is cystinosis [45], a lysosomal storage disease characterized by the abnormal accumulation of the amino acid cystine [46,47]. It is transmitted as an autosomal recessive trait and has three forms: infantile (nephropathic), late-onset (juvenile), and adult (benign). Patients with the adult form do not develop urinary problems and only experience ocular manifestations such as photophobia [46].

Acquired causes of Fanconi syndrome
Cystinosis is caused by mutations in the CTNS gene that encodes the lysosomal cystine transporter, cystinosin [46,48]. This leads to the accumulation of cystine within lysosomes and results in end-organ damage [46,48]. Renal proximal tubular cells are apparently highly susceptible to the effects of excessive accumulation of cystine, including damage to the renal proximal tubular cells and the ensuing Fanconi syndrome [35]. Various mechanisms are involved in the kidney damage observed in cytinosis including cysteinylation of protein kinase delta, which increases apoptosis of the cysteine-laden renal proximal convoluted tubule cells [49,50]; ATP depletion and inhibition of Na+ dependent transporters secondary to cysteine accumulation [50,51]; and decreased expression of megalin, cubilin, and sodium transporters at the apical surface of proximal convoluted tubule cells [50].
Dent disease is not usually considered a cause of Fanconi syndrome but it shares some features of Fanconi syndrome, such as hypophosphatemia. Dent disease is an X-linked disorder with two subtypes. Dent disease 1 is caused by mutations in the CLCN5 gene, which encodes for an endosomal hydrogen chloride (H+/Cl−) exchange transporter. Dent disease 2 is caused by mutations in the OCRL gene, which encodes for a 5-phosphatase that is involved in cellular trafficking [52]. Both subtypes of Dent disease manifest in childhood in as varying degrees of proximal tubular dysfunction, including low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, hypophosphatemia, and renal failure that eventually progresses to end-stage renal disease. Dent disease 2 tends to be less severe.
Mutations in the OCRL pathway can also lead to Lowe syndrome (also known as oculocerebrorenal syndrome), an X-linked disorder that affects the kidneys, eyes, and the brain. In Lowe syndrome, which is more severe than Dent disease 2 in terms of renal manifestations, bicarbonate wastage leads to the development of proximal RTA [53].
Acquired causes of Fanconi syndrome
Drug-induced nephrotoxicity is the most common acquired cause of Fanconi syndrome, followed by light-chain-associated Fanconi syndrome [54]. The acquired causes of Fanconi syndrome are summarized in Table 2.
Fanconi syndrome associated with drug nephrotoxicity
Anti-retroviral medications
Numerous antiretroviral medications have been implicated in Fanconi syndrome in human immunodeficiency virus (HIV)-positive patients [55,56]. Earle et al [56] described three cases in which HIV patients developed generalized tubular dysfunction with hypophosphatemia, metabolic acidosis, phosphaturia, glycosuria, and generalized aminoaciduria. Serum analysis revealed hypophosphatemia in all three, but serum bicarbonate was low-normal. Tenofovir, adefovir, and cidofovir are all nucleotide reverse-transcriptase inhibitors. Nonetheless, Fanconi syndrome has also been reported with nucleoside reverse-transcriptase inhibitors such as lamivudine, stavudine, and didanosine [57–59].
Three mechanisms of tenofovir nephrotoxicity have been suggested, including drug excretion in the proximal tubules, genetic association, and mitochondrial toxicity [60]. Proximal tubules are involved in the excretion of several drugs, including tenofovir, which enters proximal tubule cells through basolateral organic anion transporters and exits using apical transporter multidrug-resistance-associated protein 4 [61]. Didanosine uses the same organic anion transporters to enter the proximal tubular cells. When combined with tenofovir, both drugs compete for the same transporters, leading to increased renal toxicity [60].
Renal dysfunctions (including Fanconi syndrome) due to tenofovir are usually reversible with cessation of the drug, although persistent Fanconi syndrome has been reported. Data regarding tenofovir dose reduction as a strategy to ameliorate nephrotoxicity and aid in the recovery of renal function are lacking. Because of severe nephrotoxicity, tenofovir disoproxil fumarate (TDF) has been almost completely replaced by tenofovir alafenamide (TAF) for treatment of HIV infection and chronic hepatitis B virus infection in the United States. One of the reasons for this change is the fact that TAF is less nephrotoxic than TDF [62,63]. However, a case of Fanconi syndrome in a patient treated with TAF has been recently reported [63].
Several reports have been published in which didanosine is associated with the development of Fanconi syndrome in HIV-positive patients [58,59,64]. Izzedine et al [59] reported a case of Fanconi syndrome and diabetes insipidus in an HIV patient treated with didanosine among other medications. The patient was hospitalized for fatigue, dehydration with weight loss, and polyuria. Laboratory examination on admission revealed normal anion-gap metabolic acidosis hypokalemia, hypophosphatemia, and hypouricemia. A 24 hours urine collection on the second hospital day revealed a urine output of 4.5 L, heavy glycosuria in spite of normal serum glucose, aminoaciduria, and 3.5 g of mixed proteinuria [59]. After didanosine cessation, and despite the continuation of adefovir, the plasma levels of bicarbonate potassium and uric acid normalized. Urinalysis was negative for glycosuria, with a progressive reduction in proteinuria [59].
D’Ythurbide et al [64] also reported Fanconi syndrome and diabetes insipidus in an HIV patient being treated with didanosine, lamivudine, atazanavir, and ritonavir. On admission, the patient had hypophosphatemia, hypouricemia, hyperchloremic metabolic acidosis with a normal anion gap, normoglycemic glycosuria, low-molecular-weight proteinuria, and high fractional excretion of phosphate [64]. The patient recovered completely one month after discontinuation of didanosine, while treatment with the other medications was continued [64].
Ifosfamide
Ifosfamide is an alkylating agent used to treat different types of cancers in adult and pediatric populations [65,66]. A synthetic analog of cyclophosphamide, its use is sometimes limited by its adverse urological and renal toxicities, such as hemorrhagic cystitis and Fanconi syndrome. While both ifosfamide and cyclophosphamide can cause hemorrhagic cystitis, only ifosfamide is associated with Fanconi syndrome. The introduction of the uro-protective thiol compound “Mesna” (sodium 2-mercaptoethanesulfonate) has virtually eliminated urotoxicity associated with both cyclophosphamide and ifosfamide and allowed the use of higher and more frequent dosing. However, mesna has shown no preventive effect on the tubular toxicity of ifosfamide, which manifests in Fanconi syndrome [67,68]. This is likely due to insufficient delivery of mesna to the renal tubule, leading to failure to provide adequate protection of tubular glutathione from depletion by the metabolite(s) [69].
The incidence of Fanconi syndrome in treated patients is reportedly between 1.4% and 5% [67]. The toxicity of ifosfamide and the late onset of the syndrome following discontinuation have been recognized in several studies [67,70–72].
Most information on ifosfamide nephrotoxicity comes from studies in children as its use in pediatric oncology is common [70–72]. By contrast, reports of ifosfamide-related Fanconi syndrome in adult patients are scarce [73,74]. In a long-term assessment of ifosfamide-related renal toxicity in adult patients, Farry et al [75] reported a steady decline in the estimated GFR, although none of the patients progressed to end-stage renal disease.
Several studies have improved our understanding of the mechanism of renal injury due to ifosfamide. Studies in rats by Nissim et al [76] showed that chloroacetaldehyde (CAA), the active metabolite of ifosfamide, causes renal injury by inhibiting nicotinamide adenine dinucleotide (reduced) (NADH):ubiquinone oxidoreductase (Complex-1; C-I), one of the enzymes in the oxidative phosphorylation pathway. The authors demonstrated that CAA accumulates in the kidney cortex following ifosfamide treatment. Inhibition of C-I led to increased NADH and decreased NAD. Moreover, the administration of agmatine (AGM), a metabolite of arginine decarboxylation, with ifosfamide prevented these changes and raised cyclic adenosine monophosphate levels. AGM, therefore, has been suggested as a potential compound for the prevention of ifosfamide-induced tubular dysfunctions, including Fanconi syndrome [76]. Yaseen et al [77] discovered that CAA inhibits endocytosis in the rat proximal tubules. This inhibition was attributed to a decrease in ATP levels and inhibition of Vacuolar-type H+-ATPase induced by CAA.
Oxaliplatin and cisplatin
Oxaliplatin has been reported to cause both isolated proximal RTA as well as proximal RTA with Fanconi syndrome [78,79]. It was first reported in a patient being treated with oxaliplatin for colon adenocarcinoma. The patient developed hypokalemic, hyperchloremic metabolic acidosis with a normal anion gap [78]. The presence of glycosuria and low serum phosphate levels prompted the diagnosis of Fanconi syndrome [78]. In another report of oxaliplatin treatment for adenocarcinoma of the colon, the patient developed HCO3− wasting and severe hypokalemic, hyperchloremic metabolic acidosis with a normal anion gap but no other abnormalities, suggesting isolated proximal RTA [79].
Fanconi syndrome has also been described with cisplatin [80]. Cisplatin administration in mice caused a significant increase in the urinary concentrations of glucose, amino acids, and trichloroacetic acid cycle metabolites, such as pyruvate and lactate, within 48 hours of administration [81]. Aminoaciduria in cisplatin-treated mice also preceded the elevation of serum creatinine. Cisplatin inhibits peroxisome proliferator-activated receptor-alpha activity and consequently fatty acid oxidation resulting in proximal tubular cell death [81,82]. Fibrates such as bezafibrate prevent this inhibition and may be protective against cisplatin-induced proximal tubule cell death [82]. It has also been suggested that one of the possible mechanisms of cisplatin-induced proximal tubule nephrotoxicity is reduced expression and function of sodium-dependent glucose transporters [81].
Anticonvulsant therapies
Several studies have reported the development of metabolic acidosis following treatment with topiramate [83–85]. Topiramate is an anti-epileptic drug used for seizure disorders and migraine prophylaxis [83]. Sacré et al [86] reported a case in which topiramate was used for migraine prophylaxis in a patient who developed hyperchloremic metabolic acidosis with a normal GFR and positive urine anion gap (UAG). The authors reported that the patient had both proximal and distal RTA. It has been suggested that inhibition of CA II by topiramate can cause development of mixed RTA [39,87]. Winum et al [39] found that topiramate is a potent inhibitor of human CA II and XII and a medium potency inhibitor of CA IV. However, Maryanoff et al [88] found that topiramate has low activity against CA II. X-ray crystallography studies have revealed a close association between bound topiramate and the active site of CA II, a finding that is congruent with topiramate’s potent inhibitory activity against CA II [87]. The inhibition of cytosolic CA II by topiramate provides a reasonable explanation for the development of mixed RTA.
Another anticonvulsant that can rarely be a cause of Fanconi syndrome is valproic acid. A few case reports of Fanconi syndrome with the use of valproic acid have been published [89–91]. Several reports describe normalization of laboratory values after cessation of valproate [90,91]. The mechanism for this reaction is not clear, but a direct toxic effect of valproic acid on mitochondria in the proximal tubules has been suggested [92,93].
Miscellaneous
Maleic acid
In a model of Fanconi syndrome produced by administering maleic acid to rats and dogs, renal cortical Na+/K+-ATPase activity is markedly diminished [94]. The relevance of these observations to human Fanconi syndrome remains to be determined but the generalized proximal tubular dysfunction in Fanconi syndrome could be a result of a generalized defect of Na+-coupled apical membrane transporters, a defect in basolateral Na+/K+-ATPase, or metabolic disorders that lower intracellular concentrations of ATP [3].
Deferasirox
Diseases such as beta thalassemia, sickle cell disease, and myelodysplastic syndromes require long-term blood transfusions as the mainstay of treatment. Iron-chelating agents have been developed to counteract accumulation of dangerous levels of iron. The main agent, deferoxamine, requires administration as a slow, continuous subcutaneous or intravenous infusion five to seven times per week [95], which may lead to non-compliance. Deferasirox, which is administered orally using a once-a-day dosing schedule, is far more likely to ensure compliance and is therefore increasingly being prescribed by hematologists. However, nephrotoxicity is the most serious and frequent adverse effect of deferasirox treatment and can present as an acute or chronic decrease in GFR. Features of proximal tubular dysfunction may also be present [96].
Several recent case reports [97–99] and one study [100] have linked deferasirox to the development of Fanconi syndrome in patients undergoing iron-chelation therapy. In one retrospective study by Chuang et al [100], five of 57 patients with thalassemia treated with deferasirox developed Fanconi syndrome. The authors hypothesized that deferasirox is more toxic to the less-mature renal tubules in younger children when compared with the effect on the study’s adult patients, who were relatively resistant to Fanconi syndrome [100]. Renal tubular dysfunction was not related to the dose of deferasirox and dose reduction did not ameliorate tubular dysfunction. Several mechanisms have been proposed, including deferasirox accumulation in the kidney, modulation of hemodynamics by excessively rapid iron removal, and iron-dependent cell death, but an exact mechanism has yet to be elucidated [96].
Aminoglycosides
The presence of glycosuria and aminoaciduria after exposure to gentamicin has been reported in rats [101]. Several cases of patients with aminoglycoside-induced Fanconi syndrome have been reported [102–105]. Ghiculescu and Kubler [102] reported the development of Fanconi syndrome in a 53-year-old man treated for respiratory infections with gentamicin. The patient developed hypophosphatemia, hypocalcemia, hyperphosphaturia, and aminoaciduria. These electrolyte disturbances persisted until gentamicin therapy was stopped, recurred with a re-challenge, and did not correct with calcium and phosphate supplementation [102].
In most reported cases, the alteration in reabsorption predominately affected neutral amino acids and, to a lesser extent, acidic amino acids [102].
In the case of gentamicin, recent in vitro and in vivo studies performed in LLCPK1 cells, as well as in mouse kidney tissue, have shown that aminoglycoside antibiotics reduce glucose reabsorption in kidney tissue by reducing mRNA, protein expression, and function of sodium-dependent glucose transporters in the apical membrane of the proximal tubule [106].
L-cationic amino acids
L-cationic amino acids, such as L-lysine and L-arginine, have a profound inhibitory effect on proximal HCO3− reabsorption and can potentially cause proximal RTA [107].
Ingestion of L-lysine has been reported as a cause of Fanconi syndrome [108]. Several cases of Fanconi syndrome have been reported involving patients with lysinuric protein intolerance (LPI) [109,110], a rare autosomal recessive multiorgan disorder in which the renal and intestinal transport of the cationic amino acids lysine, arginine, and ornithine is defective [111]. This leads to heavy L-lysine loads on renal tubules. Most, but not all, of the symptoms of LPI have been linked to a secondary urea-cycle derangement due to impaired transport of cationic amino acids. Typically, symptoms begin after weaning, with refusal of feeding, vomiting and consequent failure to thrive [112,113]. Two major complications, pulmonary alveolar proteinosis and renal disease, are increasingly observed in LPI patients [113,114]. Interestingly, the authors did not consider the intratubular accumulation of lysine as a cause for the patient with Fanconi syndrome [109]. This is supported by the fact that, hyperlysinemia, an autosomal recessive disease characterized by increased lysine levels in the blood due to a mutation in the enzyme α-aminoadipic semialdehyde synthase, is relatively benign and does not lead to Fanconi syndrome [115]. Instead, the authors suggest that the cause of the proximal tubular dysfunction in LPI is either due to diminished resorptive area and decreased availability of transport proteins, or increased back-leak of solutes from the proximal tubular cell [110].
Apremilast
Apremilast is a phosphodiesterase 4 inhibitor used in the treatment of psoriasis and psoriatic arthritis. In a report describing a case of Fanconi syndrome in a patient treated with apremilast [116], the patient developed hypokalemia, hyperchloremic metabolic acidosis, low uric acid concentration, a positive urinary anion gap, and proteinuria two weeks after the initiation of apremilast therapy [116]. These abnormalities resolved upon discontinuation of the drug. Two months after the initial event, the patient resumed apremilast therapy, but developed similar abnormalities after 17 days. Apremilast was again discontinued and the patient recovered after [116].
Heavy metals
Heavy metals such as lead, cadmium, and mercury are reportedly associated with proximal RTA [117,118] and chronic cadmium exposure has been reported to cause Fanconi syndrome [119,120]. Cadmium accumulates in the proximal tubular cells through receptor-mediated endocytosis of metallothionein-bound Cd (Cd–MT). Cd–MT complexes are degraded in endosomes and lysosomes that release free Cd2+ into the cytosol, where it generates reactive oxygen species, triggering a cascade of damaging cellular events that can cause generalized proximal tubular dysfunction [119].
Fanconi syndrome associated with systemic disease
The acquired causes of Fanconi syndromes are usually associated with features of proximal RTA and include amyloidosis, multiple myeloma, paroxysmal nocturnal hemoglobinuria, renal transplantation (Table 2) [54,121–124]. A discussion of these individual entities is beyond the scope of this review.
Clinical diagnosis
The hallmark of Fanconi syndrome is increased excretion of substances usually reabsorbed in the PCT while their levels in the plasma are not elevated. The presence of proximal RTA usually raises the suspicion of Fanconi syndrome in patients receiving one of the causative agents.
Fanconi syndrome is usually suspected when a patient develops tubulopathy in association with one of the causes discussed previously. There is often a temporal relationship between the cause and the development of Fanconi syndrome, although exceptions exist in which the syndrome develops months or years after the introduction of the causative agent (such as tenofovir [125]). Other causes, such as apremilast, can result in metabolic derangements within a few weeks of starting treatment.
A diagnosis of proximal RTA requires establishing the presence of urinary HCO3− wastage. This is demonstrated with a high urine pH in the presence of normal or slightly reduced plasma HCO3−. Traditionally, a fractional HCO3− excretion of ≥ 15% when the plasma HCO3− is normal is said to be needed to establish the diagnosis of proximal RTA [2]. However, in the proper context, a fractional excretion of only 5% is consistent with proximal RTA because the distal and collecting tubules can reclaim some of the HCO3− “rejected” by the proximal tubule. When the fractional excretion is 15% or higher, the defect in the proximal tubule for HCO3− reabsorption must be quite severe.
As previously mentioned, patients with proximal RTA can lower urine pH to < 5.5 when the plasma HCO3− is lower than the renal threshold for HCO3− reabsorption. This is in contrast to patients with classic or type I RTA who cannot lower urine pH maximally regardless of the degree of the acidosis (Fig. 2).
Another bedside diagnostic tool is the use of the urinary anion gap (UAG) as a surrogate marker of ammonium excretion. The UAG is consistently increased (positive) in distal RTA [126–128]. By contrast, in proximal RTA and in the presence of metabolic acidosis, one would expect the UAG to be appropriately decreased (negative). However, there are limited data on the UAG in proximal RTA, and some patients with proximal RTA may not have a maximal ammonium excretion during acidosis. Accordingly, the UAG may not be appropriately reduced (negative). Data on urine ammonium and UAG in proximal RTA are clearly needed.
Urine studies, which are underutilized in clinical practice, are necessary for the diagnosis of Fanconi syndrome. There is usually increased fractional excretion of phosphate and uric acid as well as euglycemic glycosuria. Increased urinary excretion of amino acid and low-molecular-weight proteins are sensitive markers of proximal tubulopathy and often used to confirm the diagnosis of Fanconi syndrome. Urinary retinol-binding protein 4 in urine appears to be the most sensitive biomarker for proximal tubulopathy but it is not readily available as a diagnostic tool in most places [129]. The diagnosis is often confirmed by the improvement of tubulopathy following the discontinuation of the offending agent(s).
Treatment
The autosomal dominant and recessive forms of proximal RTA are usually permanent and require life-long alkali therapy. In contrast, sporadic isolated proximal RTA is transient and alkali therapy can be discontinued after several years. After discontinuation of the alkali therapy, the condition does not return. This defect has been ascribed to proximal tubular immaturity in some infants [32].
The goal of treatment in Fanconi syndrome is to stop the causative agent (when possible) and to replenish the solutes that are wasted in the urine due to the lack of proximal tubular reabsorption. Loss of glucose, uric acid and amino acids is usually asymptomatic. Loss of phosphate, however, can lead to rickets in children and osteomalacia in adults. Hypophosphatemia in Fanconi syndrome and proximal RTA can be exacerbated by vitamin D deficiency, and both phosphate and vitamin D levels should be checked and supplements administered [130].
All patients with proximal RTA should receive HCO3− supplementation. Alkali therapy is of particular importance in children to prevent growth retardation due to acidosis. Large amounts of HCO3− are usually necessary given the magnitude of HCO3− wastage in proximal RTA and sometimes Fanconi syndrome. An amount of 5 to 15 mEq/kg is usually recommended [3]. Thiazide diuretics enhance HCO3− reabsorption in the PCT and the loop of Henle by reducing extracellular volume and are used to minimize the required amount of HCO3−. Potassium levels should be monitored and supplemented when necessary, given that both HCO3− supplementation and thiazide diuresis lead to hypokalemia by enhancing K+ excretion in the collecting duct [3].
Any time a medication is suspected to be the cause of Fanconi syndrome in a patient, it should be stopped if possible and efforts should be made to find an alternative. When a suitable replacement is not present, lowering the dose should be performed, although, as mentioned previously, some cases of Fanconi syndrome are not dose-related.
Notes
Conflicts of interest
All authors have no conflicts of interest to declare.
Authors’ contributions
Daniel Batlle and Ibrahim Kashoor participated in the data collection and wrote the manuscript. Daniel Batlle and Ibrahim Kashoor participated in the study design. Daniel Batlle and Ibrahim Kashoor participated in the conception, analysis, and interpretation of data. Daniel Batlle and Ibrahim Kashoor provided intellectual content of critical importance to the work and technical support. Daniel Batlle and Ibrahim Kashoor participated in the study design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.