The insulin-like growth factor system in chronic kidney disease: Pathophysiology and therapeutic opportunities
Article information
Abstract
The growth hormone–insulin-like growth factor–insulin-like growth factor binding protein (GH–IGF–IGFBP) axis plays a critical role in the maintenance of normal renal function and the pathogenesis and progression of chronic kidney disease (CKD). Serum IGF-I and IGFBPs are altered with different stages of CKD, the speed of onset, the amount of proteinuria, and the potential of remission. Recent studies demonstrate that growth failure in children with CKD is due to a relative GH insensitivity and functional IGF deficiency. The functional IGF deficiency in CKD results from either IGF resistance due to increased circulating levels of IGFBPs or IGF deficiency due to increased urinary excretion of serum IGF–IGFBP complexes. In addition, not only GH and IGFs in circulation, but locally produced IGFs, the high-affinity IGFBPs, and low-affinity insulin-like growth factor binding protein-related proteins (IGFBP-rPs) may also affect the kidney. With respect to diabetic kidney disease, there is growing evidence suggesting that GH, IGF-I, and IGFBPs are involved in the pathogenesis of diabetic nephropathy (DN). Thus, prevention of GH action by blockade either at the receptor level or along its signal transduction pathway offers the potential for effective therapeutic opportunities. Similarly, interrupting IGF-I and IGFBP actions also may offer a way to inhibit the development or progression of DN. Furthermore, it is well accepted that the systemic inflammatory response is a key player for progression of CKD, and how to prevent and treat this response is currently of great interest. Recent studies demonstrate existence of IGF-independent actions of high-affinity and low-affinity-IGFBPs, in particular, antiinflammatory action of IGFBP-3 and profibrotic action of IGFBP-rP2/CTGF. These findings reinforce the concept in support of the clinical significance of the IGF-independent action of IGFBPs in the assessment of pathophysiology of kidney disease and its therapeutic potential for CKD. Further understanding of GH–IGF–IGFBP etiopathophysiology in CKD may lead to the development of therapeutic strategies for this devastating disease. It would hold promise to use of GH, somatostatin analogs, IGFs, IGF agonists, GHR and insulin-like growth factor-I receptor (IGF-IR) antagonists, IGFBP displacer, and IGFBP antagonists as well as a combination treatment as therapeutic agents for CKD.
Introduction
In 2002, the U.S. National Kidney Foundation's Kidney Disease Outcomes Quality Initiative (K/DOQI) clinical practice guidelines provided for the first time a working definition of chronic kidney disease (CKD), irrespective of the cause of kidney disease, based on the presence of kidney damage and/or reduced estimated glomerular filtration rate for more than 3 months. CKD is largely caused by diabetes, hypertension, glomerulonephritis, obesity, and metabolic syndrome [1], [2], [3], [4], [5], [6], [7]. Moreover, CKD is recognized as a major risk factor for end-stage renal disease (ESRD), cardiovascular disease, and cardiovascular disease-related premature death [8], [9]. CDK is fast becoming a worldwide epidemic and recognized as a major public health problems, with 26 million American adults suffering from CKD and millions of others at increased risk [10]. CKD results in complex metabolic and hormonal disturbances, particularly in the growth hormone–insulin-like growth factor–insulin-like growth factor binding protein (GH–IGF–IGFBP) axis. Perturbations in this somatotropic hormone axis are responsible for many important complications seen in CKD, such as growth retardation and catabolism, as well as disease progression. This review will focus on recent discoveries and insights into the IGF system and its therapeutic potential in CKD.
The IGF system
GH–IGF axis
GH exerts its somatotropic effects partially by stimulating the production of IGF-I in the liver through activation of hepatic GH receptor. IGF-I is transported to the main target organs via the circulation to act as an endocrine factor [11]. In addition, circulating IGF-I acts as a feedback on the somatotropic axis and suppresses the release of GH from the pituitary gland [11].
The IGF–IGFBP–IGF-rP superfamily
IGF-I, IGF-II, their cognate receptors (insulin-like growth factor-I receptor [IGF-IR] and insulin-like growth factor-II receptor [IGF-IIR]), high-affinity binding proteins (IGFBP-1 to IGFBP-6), low affinity IGFBP-related proteins (IGFBP-rP1 to IGFBP-rP7), and IGFBP proteases comprise a complex system that plays a significant role not only in somatic growth and body composition but also in diseases such as cancer, diabetes, and malnutrition by serving as endocrine, autocrine, and paracrine stimulators of mitogenesis, differentiation, survival, and cellular transformation [12].
IGF-I and -II peptides: These hormones share approximately 50% homology to insulin. They are ubiquitously expressed, highly homologous small hormone peptides of approximately 7 kDa molecular mass with multiple endocrine and paracrine/autocrine activities [12]. Most circulating IGF-I is produced by the liver and is responsible for growth and development.
IGF-I and -II receptors: IGFs interact with specific cell surface receptors, designated as IGF-I and IGF-II receptors. The IGF-I receptor is a transmembrane heterotetramer consisting of two α and two β subunits. There is approximately 60% sequence homology between the IGF-I receptor and the insulin receptor [13]. The IGF-I receptor, like the insulin receptor (IR), possesses intrinsic tyrosine kinase activity. IGF-II also binds to the IGF-I receptor but with twofold lower affinity than IGF-I [13]. The IGF-II receptor, which is identical to the cation-independent mannose-6-phosphate receptor, binds IGF-II with 500-fold increased affinity over IGF-I [14]. Most of the biologic actions of IGF-II are thought to be mediated via the IGF-I receptor [14]. The IGF-II receptor is known to function primarily as a scavenger receptor, regulating the internalization and degradation of extracellular IGF-II, thus regulating the circulating IGF-II levels.
High-affinity IGFBPs: A total of six high-affinity binding proteins have been identified, IGFBP-1 through IGFBP-6 [15], [16], [17], [18] (Fig. 1). Hepatic IGF-I circulates almost entirely bound to IGFBPs. IGFBP-3, a major IGFBP species in circulation, binds 75–90% of circulating IGF-I in a large ternary complex consisting of IGFBP-3, acid-labile subunit (ALS), and IGF. ALS and IGFBP-3 are produced in the liver as a direct or indirect effect of GH. The ALS stabilizes the IGF–IGFBP-3 complex, reduces the passage of IGF-I to the extravascular compartment, and extends its half-life [19]. It is evident that certain high-affinity IGFBPs have intrinsic hormone activity (IGF-independent actions) in addition to its actions to bind IGFs and sequester the active hormone, thereby reducing IGF biologic activity (IGF-dependent actions) [20].
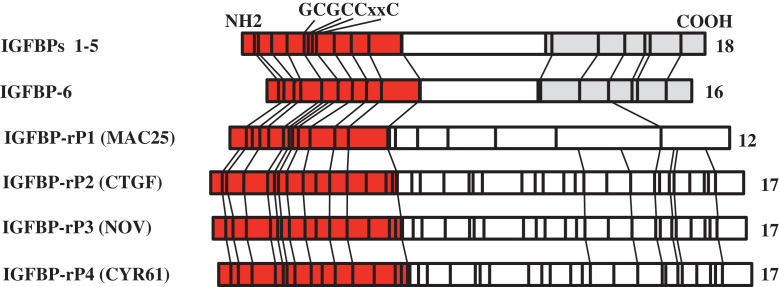
Schematic primary structures of high-affinity insulin-like growth factor binding protein-1 (IGFBP-1) to -6 and low-affinity insulin-like growth factor binding protein-related proteins (IGFBP-rPs). Numbers of conserved cysteines are indicated on the right of each structure, and locations of the cysteines are indicated as vertical lines. The conserved N- and C-terminal domains are shown as dark, stippled bars. The consensus motif GCGCCxxC is as indicated.
Low-affinity IGFBP-rPs: Several low-affinity IGFBP-rPs have been identified [12] (Fig. 1). The IGFBP-rPs are cysteine rich, and consist of four modules; an IGF binding domain, a von Willebrand factor type C repeat (involved in oligomerization), a thrombospondin type 1 repeat (involved in extracellular matrix [ECM] binding), and a C-terminal module, which may be involved in receptor binding. The IGFBP-rPs bind to IGFs with more than 100-fold lower affinity compared with the IGFBPs. The 100-fold lower affinity of IGFBP-rPs for IGFs is believed to result from loss of the C-terminal domain of IGFBPs, and both C- and N-termini are thought to be required for high-affinity binding [12]. Further, the IGFBP-rPs has actions that are predominantly independent of IGFs. In this review, we only address IGFBP-rP2 with respect to its physiologic significance and pathophysiology in interstitial fibrosis in the kidney.
IGFBP proteases: These proteases constitute another means for the regulation of IGF bioactivity and IGFBP actions. The IGF/IGFBP complex can be dissociated by proteases that cleave IGFBPs. In addition, certain free IGFBPs can also be proteolyzed by specific proteases, resulting in loss of high affinity for IGFs. To date, several proteases for IGFBPs have been described in biologic fluids and a variety of cell culture systems. These include serine proteases, prostate-specific antigen (PSA), cathepsins, and matrix metalloproteinases [21].
IGF/IGF-IR-dependent and IGF/IGF-IR-independent actions of IGFBPs
IGF-dependent actions of IGFBPs
IGFBPs are known to modulate the actions of IGFs in circulation as well as the immediate extracellular environment [16], [17]. Many IGFBPs inhibit IGF actions by binding them and preventing binding of IGFs to the IGF receptors [22], [23]. There are in vivo studies showing that IGFBPs reduce the level of free IGF, thereby inhibiting growth [24], [25], [26]. The use of an IGF analog des-(1–3)-IGF-I confirmed the sequestration mechanism in in vitro and in vivo studies. The IGF analog binds to IGF-IR and stimulates DNA synthesis, but does not bind to IGFBPs [27]. It has also been reported that a number of IGFBPs (e.g., IGFBPs-1, -3 and -5) stimulate IGF actions in a variety of cell types. A single IGFBP can enhance or inhibit IGF actions, depending on a number of variables, such as cell type, IGFBP concentration, and posttranslational modifications [17], [18]. These include proteolysis of IGFBPs, phosphorylation of IGFBPs, and binding of IGFBPs to ECM proteins. IGFBP proteases are capable of cleaving IGFBPs into forms with significantly reduced or no affinity for IGFs. Some IGFBPs (e.g., IGFBP-1 and -5) on phosphorylation exhibit increased affinity to IGF-I, whereas some remain unaffected by phosphorylation [28], [29]. IGFBPs, especially IGFBP-3 and IGFBP-5, have been reported to bind to the cell surface or ECM and show less affinity [16], [17], [18].
IGF-independent functions of IGFBPs
IGFBPs have a multitude of functions from prolonging the half-life of IGFs to playing the role of growth factors independent of IGF/IGF-R. IGF/IGF-IR independent effects of IGFBPs occur either by IGFBP binding to its binding partners on the cell surface, in cytoplasm or nucleus in certain cell types. IGF-independent actions of IGFBPs include effects on cell migration, cell growth, and apoptosis. IGFBP-1 has been reported to increase cell migration through binding to the fibronectin receptor [30]. Recent study also demonstrates an IGF-independent effect of IGFBP-5 in osteoblasts from IGF-I knockout mice [31]. IGFBP-3 has important IGF-independent effects in vitro and in vivo [32]. IGFBP-3 is one of the genes transcriptionally activated by the tumor suppressor gene p53, whose function is to induce cell cycle arrest or apoptosis and thus prevent the propagation of damaged cells [33], [34]. Several lines of evidence reveal that IGFBP-3 exerts proapoptotic and antiproliferative IGF/IGF-IR independent actions through IGFBP-3 receptor, IGFBP-3 interacting proteins, and nuclear association. The concept of IGF-independent action of IGFBP-3 was first demonstrated in breast cancer cells due to cell surface binding between IGFBP-3 and cell surface proteins typical of receptor ligand interactions [35]. A recent article reported the identification of a novel death receptor (IGFBP-3R) that specifically binds IGFBP-3 and mediates a variety of biologic functions of IGFBP-3 in normal and malignant cells [36], [37], [38], [39]. Some of the additional binding partners of IGFBP-3 are humanin, RNA polymerase II binding subunit 3 (Rpb3), and GalNAc-T14. Humanin is a peptide that inhibits neuronal cell death induced by mutant genes in Alzheimer disease [40]. Rpb3 aids the recruitment of the polymerase complex to specific transcription factors leading to the role of IGFBP-3 in the modulation of gene transcription [41]. IGFBP-3 has a nuclear localization sequence and studies show that it binds to nuclear retinoid X receptor (RXR), which is involved in physiologic function of thyroid hormone and steroid hormones, embryonic development, apoptosis, and homeostasis [42], [43], [44]. RXR heterodimerizes with Nur77, a nuclear receptor transcription factor, thereby enhancing its DNA binding ability and regulating apoptosis in various cancers [45], [46]. In the presence of IGFBP-3, Nur77 translocates to the nucleus and initiates apoptosis [47]. Many in vitro, in vivo, and epidemiologic studies suggest that IGFBP-3 may have a protective effect in cancer patients [48]. This is also emphasized by the fact that higher IGF-I and lower IGFBP-3 levels are associated with an increased risk in cancers such as prostate, colorectal, and breast. In addition, significant circulating levels of IGFBP-3 in prostate cancer correlate with slower rate of tumor progression and better therapeutic response [32], [49].
Apart from evidence supporting antitumor effect of IGFBP-3 in a variety of cancers, evidence exists to suggest that IGFBP-3 can also act as an antiinflammatory factor, thereby inhibiting physiologic manifestations of asthma such as airway inflammation and hyperresponsiveness [38]. This involves activation of IGF-independent IGFBP-3 signaling and cross-talk with nuclear factor-kappa B signaling.
Renal actions of the IGF system
The IGF system is expressed in a complicated manner within the kidney and has profound effects on kidney growth, structure, and function [50], [51], [52], [53]. However, less information is available about the expression of the IGF system in human kidney. A composite of the location of IGFs, their receptors, and high- and low-affinity IGF binding proteins in the human kidney is provided in Fig. 2. IGF-II is synthesized by the glomerular and peritubular vasculature as well as the interstitium, whereas IGF-I is expressed to a lesser extent in the glomerular vasculature [54], [55], [56]. Both IGF-I and -II receptors are present in all locations of the human kidney. This IGF-IGFR expression pattern suggests a possible autocrine/paracrine action of IGFs, in particular IGF-II in the glomerulus in addition to their endocrine actions. The IGFBPs and IGFBP-rPs are expressed in various segments of the kidney. IGFBP-1, -2, -4, and -5 and IGFBP-rP1, -rP2, and -rP4 are produced in the glomerulus predominantly. On the other hand, expression of IGFBP-3 and -6, and IGFBP-rP3 shows cortical predominance [54], [56], [57]. Although the current picture of the expression profile of the IGF system in the human kidney is incomplete, heterogeneous expression of IGFBPs and IGFBP-rPs with varying abundance implies site-specific IGF-dependent and/or IGF-independent actions of IGFBPs and IGFBP-rPs in the human kidney.
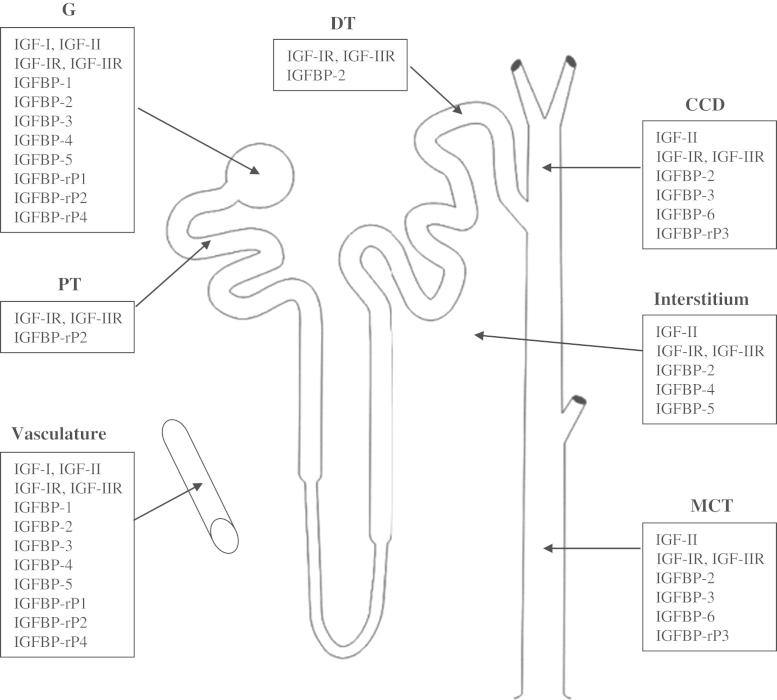
Insulin-like growth factor (IGF) system gene expression in the human kidney. A comprehensive expression profile of the members of the IGF system in human kidney is depicted in this figure. However this figure does not represent a complete profile.
CCD, cortical collecting duct; DT, distal tubule; G, glomerulus; MCT, medullary collecting duct; PT, proximal tubule.
Physiologic significance of the endocrine-derived IGFs and IGFBPs has been extensively studied in animals, in particular using transgenic (TG) mice. Administration of IGF-I to animals promotes renal growth through a process of cellular hypertrophy and hyperplasia, and induces a rapid increase in renal blood flow and glomerular filtration together with a reduction in renal vascular resistance [58]. In addition, overexpression of IGF-I in TG mice induces renal and glomerular hypertrophy [59], [60], [61]. However, no renal developmental abnormalities have been reported in newborn IGF-I knockout (KO) mice [62], [63]. Although 95% of IGF-I KO mice die immediately after birth, the mice that survive are smaller than their wild-type littermates and the weight of their kidneys is proportionally lower, along with reduced glomerular size and number of nephrons, suggesting that IGF-I plays a critical role in number of nephrons and enhances function in developing kidneys [64]. Kidney weight is decreased in two hepatic IGF-I deficiency models, probably as a result of the decreased serum IGF-I levels, because systemic delivery of IGF-I has been shown to increase kidney size [65], [66]. On the other hand, kidney weight is increased in IGF-II TG mice [67], [68]. IGFBP-1 TG mice display a small reduction in kidney weight, although proportional to body weight [69], [70], and develop glomerulosclerosis without glomerular hypertrophy [71]. Infusion of IGFBP-1 into GH-deficient dwarf mice stimulates renal but not body growth [72]. However, excessive levels of IGFBP-1 appear to have an adverse effect on the kidney, as IGFBP-1 TG mice have a reduced number of nephrons and develop glomerulosclerosis [73], [74]. IGFBP-2 overexpression inhibits GH-stimulated body and kidney growth in GH TG mice [75]. Kidney growth is disproportionately retarded by IGFBP-3 overexpression, most likely by inhibiting IGF action [76]. These data clearly demonstrate IGF-dependent inhibitory effect of IGFBPs in vivo.
It is also evident that in addition to GH and IGFs in circulation, locally produced IGFs, high-affinity IGFBPs and low-affinity IGFBP-rPs may also affect the kidney. Infusion of IGF-I directly into the renal parenchyma results in hypertrophy and hyperplasia of the medullary thick ascending limb (mTAL) and distal tubule whereas proximal tubules are not affected [77], along with minimal IGF-IR gene expression in proximal tubules [78]. IGF-I produced in the mTAL may play a trophic role in conditions associated with increased workload, such as unilateral nephrectomy and high-protein diet. In vitro, IGF-I is a potent mitogen for cultured glomerular mesangial cells and stimulates the production of ECM by mesangial cells [79], [80], [81]. IGFBP-3 alone inhibits tubular cell DNA synthesis and induces apoptosis in an IGF-dependent and -independent manner [82], [83]. This IGF-independent induction of apoptosis appears to be in part through increase of oxidative stress in porcine proximal tubular epithelial cells [84]. IGFBP-3 also mediates tumor necrosis factor-α (TNF-α)-induced mesangial cell apoptosis [85]. Furthermore, IGFBP-3 plays an important role in the development of podocyte apoptosis by modulation of transforming growth factor-β (TGF-β) and bone morphogenetic protein-7 (BMP-7)-induced proapoptotic- and antiapoptotic signals [86]. IGFBP-5 also exhibits IGF-independent actions. IGFBP-5 is able to mediate mesangial cell migration through activation of cdc42, and laminin421 binding to α6β1-integrin [87]. With respect to low-affinity IGFBP-rPs, IGFBP-rP2/connective tissue growth factor (CTGF) induces renal fibrosis in a TGF-β-dependent and -independent manner [88], [89].
Limited information is available regarding physiologic significance of the IGF system in the human kidney. IGF-I increases renal blood flow and growth factor receptor (GFR) and causes sodium retention and volume expansion [90]. Sodium retention occurs as a consequence of a direct effect on renal tubules, due to stimulation of renin release and suppression of atrial natriuretic peptide secretion [91]. IGFBP-rP2/CTGF, a potent regulator of cell proliferation, migration, and matrix production, is expressed in the immediate precursors of glomerular visceral and parietal epithelial cells in the comma- and S-shaped stages, but not in earlier stages of nephron development [92]. It is also present on precursors of mesangium and glomerular endothelium, suggesting a possible role in normal glomerulogenesis and maintenance of glomerular structure and function at adult age. In addition, IGFBP-rP2/CTGF promotes the transdifferentiation of human renal tubular epithelial cells toward myofibroblasts in vitro, suggesting a critical role of IGFBP-rP2/CTGF in interstitial fibrosis [93], [94].
The IGF system in CKD
Ample studies indicate that CKD is associated with several derangements in the GH–IGF–IGFBP axis. Perturbations in this somatotropic hormone axis are responsible for many important complications seen in CKD, such as growth retardation and catabolism, as well as disease progression. Circulating levels of GH are a balance between the production rate and metabolic clearance. GH synthesis may be reduced, normal, or even increased in CKD [95]. GH secretion is normal or low in late prepubertal and pubertal children with chronic renal failure (CRF) [96]; whereas GH secretion rates are increased with increased GH burst frequency in prepubertal children and in adults with ESRD [96], [97], [98]. The release of GH in response to GH-releasing hormone is increased in adults [99], whereas in children, the mid- to-late adolescent increase in GH production fails to occur in CKD [96]. The metabolic clearance rate of GH is obviously decreased in advanced CKD [100]. Overall, plasma GH levels are normal or increased in patients with CKD. Growth retardation is a significant complication in children with CKD and occurs even though serum GH levels are normal or even elevated, reflecting a state of acquired GH resistance.
With respect to circulating IGF-I, it has been demonstrated that serum IGF-I levels are normal or low in children with advanced CKD [101], whereas, in adult patients, serum IGF-I levels are reduced only in patients with cachexia [102]. The rate of IGF-I production is reduced in children with uremia [103], but appears to be normal in adults with ESRD [104], [105]. Insensitivity to IGF-I is an important cause of GH resistance, and this appears to arise because of the accumulation of circulating IGFBPs that are normally cleared through the kidney. Serum levels of IGFBP-1, -2, -4, and -6 are elevated. With serum IGFBP-3, immunoreactive IGFBP-3 levels are elevated because of an increase of immunoreactive IGFBP-3 fragments, which have reduced IGF-I affinity, whereas intact IGFBP-3 levels are not elevated [106], [107], [108], [109]. Therefore, the bioavailability of IGF-I in CKD is compromised because of increased levels of IGFBP-1, -2, -4, and -6 [110], [111], [112]. Furthermore, as a result of increased proteolysis of IGFBP-3, less IGF-I circulates as the 150-kDa ternary complex normally comprising IGF-I, intact IGFBP-3, and ALS. Consequently, effective delivery of IGF-I to its sites of action is reduced. Thus, IGF bioavailability is significantly reduced in CKD due to increased production and reduced clearance of IGFBPs with impaired kidney function, which results in decreased IGF bioactivity of CKD serum despite normal total IGF levels. It is also conceivable that tissue IGF-I resistance may arise due to altered local IGFBP production and accumulation. It has been suggested that administration of an inactive IGF-I analog might be used to displace IGF-I from IGFBPs in CKD and thus restore IGF-I bioactivity [113]. It is of note that the functional IGF deficiency in nephrotic syndrome (NS) appears to result from low levels of serum IGFBPs as well as serum IGFs due to increased urinary losses of serum IGF–IGFBP complexes [114]. Some studies show that in children with NS, serum IGFBP-3 returned to normal levels and IGF-I was still lower in the remission stage [114], whereas others indicate that both IGF-I and IGFBP-3 returned to normal levels [115]. It is evident that serum IGF-I and IGFBPs are altered with different stages of CKD, the speed of onset, the amount of proteinuria, and the potential of remission.
It is known that patients with diabetes and microalbuminuria also present with severe alterations in the GH–IGF–IGFBP system, with increased IGF-I renal levels and IGFBP-3 protease activity, increased excretion of bioactive GH, IGF-I, and IGFBP-3, but decreased circulating intact IGFBP-3 levels [116]. Patients with type 1 diabetes have an increased level of circulating GH and less high-affinity GH-binding protein, the soluble ectodomain of the GH receptor, which indirectly indicates a decrease in the number of GH receptors in these patients [117]. This leads to a state of GH resistance, and thus a decrease in the levels of IGF-I and IGFBP-3 [118], [119], [120]. In contrast, IGFBP-1 levels are significantly higher in patients with diabetes and microalbuminuria than in healthy adolescents [121], [122]. IGFBP-1 serum concentration is inversely related to insulin levels [122]; therefore, the relative portal insulinopenia in patients with diabetes is associated with increased IGFBP-1 levels and consequent reduction of IGF-I bioavailability [123]. In children and adolescents with diabetes there is an association between urinary IGF-I and kidney volume as well as microalbuminuria, suggesting a role of IGF-I in the pathogenesis of diabetic nephropathy (DN) [124]. A longitudinal assessment of IGF-I levels in young patients with childhood onset type 1diabetes shows that patients developing microalbuminuria have lower total and free IGF-I levels than control patients [125]. Adolescents with diabetes and microalbuminuria have lower IGFBP-3 serum concentrations, but higher levels of urinary IGFBP-3 than control patients [121], [122]. This increased urinary IGFBP-3 is related to incipient nephropathy, and not simply to poor metabolic control. Indeed, adolescents with diabetes and microalbuminuria show higher levels of urinary IGFBP-3 present as both intact form and fragments, and even when compared with patients without microalbuminuria, was matched for metabolic control [126], [127], [128]. Because IGFBP-3 has the ability to induce mesangial apoptosis in the context of high ambient glucose or TNF-α in the kidney [129], it has been suggested that DN is associated with IGFBP-3 proteolysis in urine, and the derived IGFBP-3 fragments may retain biologic activity and contribute to increased albumin excretion through increased apoptosis of glomerular epithelial and endothelial cells [128]. Table 1 summarizes the published data on the GH–IGF–IGFBP profiles in patients with CDK and DN.

GH–IGF–IGFBP Axis Profile in Serum and Urine of Patients with CKD and DN
It has also been reported that IGFBP-rP-2/CTGF, a direct mediator of TGF-β-induced fibrosis, show a significant increase of serum levels in patients with DN and correlated with albuminuria, creatinine clearance, duration of diabetes, and glycemic control [130]. In addition, in patients with diabetes and microalbuminuria or DN, urinary IGFBP-rP2/CTGF levels are increased [131], [132] and significantly correlated with urinary albumin excretion, and glomerular filtration rate [130]. A recent prospective study, with a 12.8-year follow-up period, performed in 198 adult patients with type 1 diabetes, has shown that plasma IGFBP-rP2/CTGF is an independent predictor of ESRD and mortality in patients with DN [133]. In a manner similar to TGF-β, IGFBP-rP2/CTGF present in the renal tubular fluid may act directly on tubular epithelial cells and induce the expression of ECM proteins, thereby causing tubulointerstitial fibrosis [134].
Patients with active glomerulosclerosis (FSGS) excrete high amounts of podocalyxin- positive cells as well as IGFBP-1 and -3 [135]. Furthermore, IGFBP-1 and -3 are upregulated in response to TGF-β and bradykinin in human podocytes and microvascular endothelial cells. These findings suggest that the local expression of IGFBPs, in particular IGFBP-1 and IGFBP-3 in podocytes and endothelial cells, might contribute to the pathogenesis of glomerular disease. In addition, IGFBP-rP2/CTGF appears to be abundant in FSGS and is associated with development of severe FSGS, glomerulonephritis, glomerulosclerosis, hypertensive nephrosclerosis, and kidney allograft fibrosis [92], [136], [137], [138].
Therapeutic use of the IGF system in CKD
Prevention and treatment of CKD has been based on early and aggressive treatment of hypertension. Disturbances of the GH–IGF–IGFBP axis in CKD have suggested therapeutic roles for both inhibition, as well as stimulation, of that axis in CKD. Experimental data suggest that antagonists of the GH/IGF-I system, including somatostatin analogs, GHR and IGF-IR antagonists as well as antagonists of downstream factors activated by GH/IGF-I, such as the angiotensin-converting enzyme and AGE systems, may have beneficial effects on the diabetic kidney in animal models [139], [140], [141]. A novel somatostatin analog PTR-3173, which exerts a prolonged inhibitory action selectively on the GH-IGF axis, but not on insulin secretion, shows blunted renal/glomerular hypertrophy, albuminuria, and glomerular filtration rate in the nonobese diabetic (NOD) mice, presumably through prevention of renal IGF-I accumulation [139]. Furthermore, a specific GH receptor antagonist (GHRA), G120K-PEG, exhibits normalization of the diabetes-associated renal hypertrophy and glomerular enlargement in mice with diabetes [121], [141]. Most importantly, this GHRA lowers the diabetes-induced rise in urinary albumin excretion. In addition, late intervention with GHRAs, alone or in combination with angiotensin-converting enzyme inhibitors in NOD mice with manifest renal changes, shows regression in some diabetes-associated renal changes (e.g., urinary albumin excretion and renal hypertrophy) [141]. These promising preclinical data strongly suggest that GHR blockade is a viable, new modality for treating or preventing diabetic renal complications.
On the other hand, the administration of IGF-1 has been reported in a number of small studies to show some beneficial effects on CKD and ESRD. IGF-I improves renal function after acute renal failure [142] and in CRF [143], [144]. IGF-I administration results in an improvement in renal function and an increase in kidney size over 4 days in patients with advanced CRF [145]. In a second, prolonged study with ESRD, using the same dose of IGF-I, it results in a modest increase of GFR after 4 days. However, some patients treated with IGF-I for 13 and 27 days showed no long-term effect on insulin clearance or p-aminohippurate clearance, which might be attributed to administration of the large dose of IGF-I [146]. Further studies indicate that administration of a half amount of IGF-I (50 μg/kg/day) intermittently (4 days on treatment, 3 days off treatment) can improve renal function to that achieved by dialysis [133]. These studies support a potential role of IGF-I in CKD to delay the need for dialysis. Because CKD presents a state of IGF-I resistance due to elevated serum IGFBPs, it has been suggested that administration of an inactive IGF-I analog such as Leu24,59,60Ala31–IGF-I and Leu24Ala31–IGF-I, which retain affinity for binding to the IGFBPs but show no activity on the IGF receptor, might be used to displace IGF-I from IGFBPs in CKD and thus restore IGF-I bioactivity [147], [148]. Indeed, these IGF displacers produce IGF-I-like effects including increased kidney size and markers of kidney function as well as stimulated whole body weight gain and bone growth in two animal models of IGF-deficiency [113], [148]. In addition, IGFBP-1 could be an attractive specific target for displacement in CRF, because there is evidence that IGFBP-1 may have an important role in inhibiting serum IGF-I bioavailability [149]. Particularly when phosphorylated, IGFBP-1 has a higher affinity for IGFs than membrane-bound IGF receptors and the high levels of IGFBP-1 in CRF are negatively correlated with both linear growth and GFR. Furthermore, IGFBP-1 levels normalize after a successful renal transplantation [149]. In this respect, novel peptides that show remarkable specificity for IGFBP-1 have been discovered [148], and further studies would warrant their application as novel therapeutics in CRF.
DN, one of the major microvascular complications of diabetes, is characterized by excessive collection of ECM with thickening of glomerular and tubular basement membranes and increased amount of mesangial matrix, which ultimately progresses to glomerulosclerosis and tubulointerstitial fibrosis. It has been identified that one of the common characteristics of glomerulosclerosis, tubulointerstitial fibrosis, and arterial media hypertrophy lesions of hypertensive nephrosclerosis, is a significant increase of IGFBP-rP2/CTGF tissue expression, which is associated with a concomitant increase in IGFBP-rP2/CTGF in serum and urine [136]. Recent studies further demonstrate that suppression IGFBP-rP2/CTGF results in successful inhibition of proteinuria and subsequent suppression of genes involved in mesangial matrix expansion in mouse models of type 1 diabetes, providing further support for a pathogenetic role of IGFBP-rP2/CTGF in DN [150]. These findings identify IGFBP-rP2/CTGF as a promising intervention for renal fibrosis. Indeed, FG-3019, a fully human monoclonal neutralizing immunoglobulin G antibody against IGFBP-rP2/CTGF, has been developed by FibroGen (San Francisco, CA, USA) for the treatment of fibrotic diseases and diabetic complications. Recent clinical phase 1 studies demonstrate the safety and efficacy of this antibody in patients with diabetes and microalbuminuria [151]. It is conceivable that IGFBP-rP2/CTGF would become an effective and safe therapeutic option for the treatment of progressive renal fibrosis in the near future.
Conclusion
During the past decade there has been a steady progress in our understanding of the role of the GH–IGF–IGFBP axis in the maintenance of normal renal function and the pathogenesis and progression of CKD. Ample evidence also indicates that GH and IGF-I are involved in the pathogenesis of DN and growth failure in children with CKD. It appears that not only GH and IGFs in circulation, but also locally produced IGFs, high-affinity IGFBPs, and low-affinity IGFBP-rPs alter with different stages of CKD, thereby resulting in dysregulation of the endocrine, paracrine, and autocrine effects of the GH–IGF–IGFBP axis. However, information regarding the impact of the GH–IGF–IGFBP axis in different stages of CKD and its corresponding complications is limited. A holistic understanding of the GH–IGF–IGFBP axis in the multifaceted nature of CKD could provide an opportunity to develop new therapeutic strategies to improve the management of the patient with this devastating disease.
Conflict of interest
The author has nothing to declare.