


Introduction
Focal segmental glomerulosclerosis (FSGS) is a pathological diagnosis that has been used to describe renal biopsies from patients with widely varying clinical presentations and etiologies. Patients may present with or without nephrotic syndrome, may or may not have remission of proteinuria after treatment with corticosteroids, and may experience slow or rapid decline in glomerular filtration rate. It is often not possible to determine whether an individual has FSGS or a more benign form of idiopathic nephrotic syndrome, such as minimal change nephrotic syndrome (MCNS), at the time of presentation. This fact, and the common practice of deferring renal biopsy until the patient has proven that they are resistant to treatment with corticosteroids, has led to some confusion in interpreting reports regarding the etiology, treatment, and outcomes in patients with nephrotic syndrome. Studies of both familial and sporadic cases have revealed that glomerular injury in a focal and segmental pattern is common to disorders that affect podocyte junctions, change the cytoskeleton or attach to the extracellular matrix, alter ion channels in ways that increase the energy demands of cells, or impair mitochondrial function. Viral infection and toxins have also been identified in FSGS. Finally, as exemplified by patients who experience recurrent proteinuria and glomerular disease after renal transplantation, circulating substances that interact with podocytes may cause FSGS. Experimental studies have identified podocyte mechanisms that protect or limit damage from exogenous stressors.
In this review, we will briefly summarize the major genetic abnormalities in familial FSGS and touch upon the potential role of viral infection. We will discuss several agents that have been proposed as important causes of proteinuria in FSGS, and examine strategies to remove these substances or prevent their accumulation. We will also discuss therapies that may enhance glomerular defenses and protect the kidney from the cumulative injury responsible for progression to renal failure.
Clinical course and therapy
FSGS has a highly variable course; some of this variation may occur because of the diversity of etiologies, variation in host or environmental factors or due to attempts at therapy. Few trials have included segregation by genetic testing, age or renal function at diagnosis or severity of proteinuria, and thus many do not offer the specific information required to individualize therapy. No randomized controlled trials of sufficient numbers of patients are available to provide high quality information to guide therapy of FSGS in either native kidneys or renal allografts.
Current therapy results in full or partial remission in only about 50% of patients. Treatments that have been employed include corticosteroids with or without cyclophosamide [1], cyclosporine A [2], [3], tacrolimus [4], [5], mycophenolate mofetil [6], rituximab [7], [8] and adrenocorticotropic hormone [9]. If proteinuria can be reduced by these agents or by nonspecific therapies such as angiotensin converting enzyme inhibitors, angiotensin receptor blockers, aldosterone inhibitors and/or lipid-lowering agents, the progression of renal dysfunction is slowed [10]. Patients who are young at the time of diagnosis and those of any age who progress rapidly to end-stage renal disease have an increased incidence of post-transplant recurrence of proteinuria and FSGS [11].
Regardless of the etiology of FSGS, current and proposed therapies include three general strategies:(i) identifying and reversing the primary cause of renal injury; (ii) decreasing proteinuria by interventions relating to hemodynamic parameters and/or glomerular cellular responses; and (iii) slowing renal scarring by the action of nonspecific agents that target pathways for inflammation or fibrosis. Current research studies using animal models and clinical trials are aimed at identifying novel therapies that will address each of these strategies.
Genetic causes of FSGS
The discovery of nephrin deficiency as the cause of congenital nephrotic syndrome [12] has led to the intensive study of podocyte proteins and function and the identification of additional gene alterations in proteinuric renal disease and glomerulosclerosis. In Finnish nephropathy, nephrin, the major protein of the slit-pore junction, is not expressed and slit-diaphragms are absent. The majority of gene mutations that have been identified and result in nephrotic syndrome and FSGS alter podocyte proteins related to the slit-junction complex or to control of the actin cytoskeleton [13]. Abnormal podocin and ╬▒-actinin 4 are the most common, while mutations in Neph 2, CD2AP, podoplanin, inverted formin-2, phospholipase C╬Ą1, and others affect a smaller number of families and individuals. In addition, an abnormal ion channel (TRPC 6) that causes enhanced and prolonged calcium signaling has been discovered in several families with late onset FSGS [14]. Disorders of mitochondrial function and other genetic disorders are also associated with FSGS. Clinical presentation provides clues to the classification of different disorders and genetic testing for the more common mutations is commercially available. Interpretation of genetic results is complicated by the fact that many novel mutations are being identified and their relationship to disease in an individual patient cannot always be determined. Some general guidelines for differentiating between common disorders are shown in Table 1.
Viruses and nephrotoxins in FSGS
Glomerular injury, proteinuria, and progressive renal failure were identified in patients with HIV early in the study of HIV/AIDS. HIV-associated nephropathy (HIVAN) may occur even prior to the expression of HIV antibodies, and may be reversed with highly active antiretroviral therapy [15]. More often, HIVAN is detected in patients with established disease and may progress despite successful therapy. The most common histological lesions in HIVAN are collapsing glomerulopathy and microcystic tubular lesions, but other glomerular lesions may occur in individual patients. Several accessory proteins, specifically Negative Factor (NEF) and Viral Protein R (VPR), may be sufficient to cause HIVAN as shown in animal models expressing nontransmissible virus [16].
Parvovirus has also been shown in the podocytes of some patients with FSGS [17]. The relevance of this infection to idiopathic FSGS is not fully understood. It is possible that other viral infections may also lead to FSGS through their effects on podocytes.
Toxins have been used to cause proteinuria and FSGS in experimental animals. Agents employed include Adriamycin and puromycin aminonucleoside, which have been used in many studies to induce nephrotic syndrome or glomerulosclerosis [18], [19]. Puromycin aminonucleoside alters the podocyte cytoskeleton and results in detachment. Bisphosphonates also act as podocyte toxins and produce collapsing glomerulosclerosis in humans [20]. Toxicity appears to be most common following parenteral administration of rather high doses in patients with renal insufficiency. Dosing regimens have been modified to decrease risk in patients with impaired glomerular filtration rate.
Circulating factors
Circulating factor(s) in idiopathic FSGS and in post-transplant recurrence
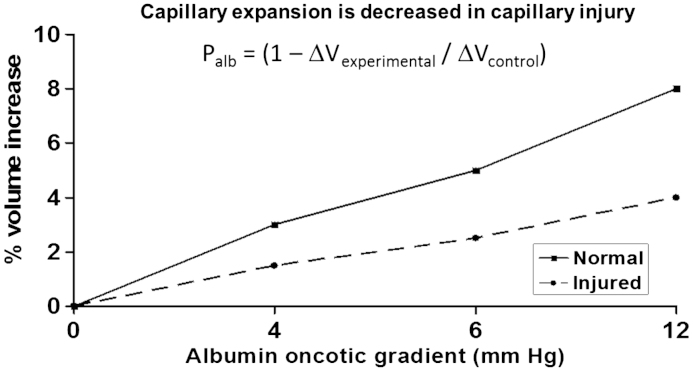
Our studies of glomerular permeability using isolated glomeruli have permitted us to test the immediate effects of a number of substances that have been implicated as mediators of injury by studies of intact animals. The assay that we developed and have used in these studies is based on the fact that transcapillary filtration can be induced by creating an oncotic gradient across the capillary wall. In our standard protocol, glomeruli are isolated in isotonic medium containing 5% bovine serum albumin and observed prior to and after a change of medium to 1% bovine serum albumin [21]. The change in bathing medium results in an oncotic gradient and causes net fluid flow into the glomerular capillaries. Capillaries expand and the entire glomerular image increases in size. Net fluid flow is proportional to the effective oncotic gradient that, in turn, is the product of the reflection coefficient of the solute (Žā) and the chemical oncotic gradient (╬ö╬Ā). In the case of normal glomeruli, Žā for albumin is one and the effective oncotic gradient equals ╬ö╬Ā. When the permeability barrier is damaged, Žā decreases, the effective gradient is diminished, and the increase in glomerular size (╬öV) is reduced proportionally. With the most severe damage, the oncotic gradient is completely ineffective, no net fluid flow occurs, and there is no capillary or glomerular expansion (╬öV=0; Žāalb=0). We have defined albumin permeability, Palb, as (1ŌĆōŽāalb). When constant conditions are used, Palb can be calculated as 1ŌĆō((╬öVexperimental)/(╬öVcontrol)). Palb is a dimensionless parameter that increases from a normal value of zero to a maximum of one with increasing severity of injury. The relationship between the oncotic gradient applied and glomerular volume increase and the calculations used are shown in Fig. 1.
We have shown that a medium containing serum or plasma from patients with recurrent FSGS and from other FSGS patients with proteinuria that is resistant to therapy increases Palb within 2ŌĆō4 minutes of incubation [22]. We used a 50-fold dilution for most experiments (2% vol/vol in isotonic medium). The increase in Palb is prevented by a variety of substances, including normal plasma [23], eicosanoids (20-hydroxyeicosatetraenoic acid, 20-HETE, and 8,9-epoxyeicosatrieneoic acid, 8,9 EET) [24], [25], cyclosporine A [26] and phosphatase inhibition [22] as shown in Table 2. The fact that numerous unrelated substances each provide glomerular protection indicates the complexity of the processes that are active in altering the glomerular permeability barrier.
MCNS has been thought to be related to a humoral factor derived from T-cells that damages the glomerular permeability barrier. Candidate permeability factors in MCNS include vascular permeability factor [27]and hemopexin [28]. Vascular permeability factor is synthesized by concanavalin A-stimulated T-lymphocytes from patients with nephrotic syndrome. It acts on both systemic capillaries and on the glomerular permeability barrier. Its secretion is enhanced by interleukins (ILs) IL-2, IL-15, IL-12, and IL-18, and is inhibited by transforming growth factor beta 1 [29], [30].
Hemopexin is a protease that appears to alter the function of both glomerular endothelium and podocytes. In cultured podocytes, it activates protein kinase B and Ras homolog gene family, member A (RhoA), and induces nephrin-dependent reorganization of the actin cytoskeleton [31], [32]. In glomerular endothelial cell monolayers, it reduces endothelial glycocalyx and increases albumin diffusion. [33]. Hemopexin injection in rats causes proteinuria and glomerular foot process fusion [34]. It is found in the urine of children with steroid-responsive nephrotic syndrome during relapse and disappears during remission.
A second substance, Cluster of Differentiation 80 (CD80), has been proposed as a urinary biomarker to distinguish between MCNS and FSGS [35]. CD80 appears to arise from podocytes. Its expression by podocytes and shedding into urine appears to be a manifestation of cellular response to injury rather than the cause of injury. Urinary CD80 is not increased in FSGS.
Circulating permeability factors in FSGS
We have carried out studies of serum and plasma from patients with FSGS for more than 20 years. Briefly, we have found that serum, plasma, or plasma fractions increase Palb during in vitro studies and cause proteinuria and albuminuria in experimental models. We have tentatively identified an IL-6 family cytokine, cardiotrophin-like cytokine-1 (CLC-1), as a cause of glomerular injury [36]. Details of our observations are given below. Other investigators have recently identified soluble urokinase receptor (suPAR) in the plasma of patients with recurrent FSGS [37]. They postulate that suPAR may be a cause of proteinuria in FSGS. This suggestion is based on the findings that suPAR concentrations are elevated in the pretransplant sera of patients with subsequent FSGS recurrence, that urokinase receptor signaling in podocytes leads to foot process effacement and urinary protein loss via a mechanism that includes lipid-dependent activation of ╬▒v╬▓3 integrin, and that transient expression of suPAR in mice leads to proteinuria and glomerular injury. Questions arise concerning the relationship between suPAR and the etiological FSGS factor, since elevated suPAR is also found in other conditions including diabetes mellitus, atherosclerosis, cancer, and inflammatory conditions [38], [39], and because levels are also elevated in uremia due to other causes [40]. The relationship between suPAR and the FSGS ŌĆ£factorŌĆØ that we have been studying is unknown.
In our studies of circulating substances in FSGS, we have focused on patients who have recurrent FSGS after renal transplantation. Proteinuria recurs following initial renal transplantation in about 30% of patients whose underlying diagnosis is FSGS [41]. Recurrence is much more common in patients with a history of allograft loss due to recurrence, and has been reported to exceed 85%. Risk factors for recurrence include young age (i.e. children and young adults), rapid course of primary disease (i.e. renal failure within 3 years of diagnosis of primary disease), previous recurrence in an allograft, and elevated permeability activity in in vitro assay [11]. Renal allografts are often biopsied to establish a diagnosis in the setting of recurrent proteinuria. Recurrent FSGS, however, is the presumptive diagnosis in patients with native kidney FSGS and proteinuria in the early post-transplant period. Biopsies in the first hours after proteinuria onset may have no morphological changes, even on electron microscopy [42]. Subsequent biopsies may show only foot process effacement, but glomerulosclerosis eventually appears unless a remission can be induced. Premature allograft loss after recurrence is common [43], [44].
Early recurrence of FSGS in renal allografts provides strong evidence that the patientŌĆÖs disease arises from a circulating substance. We have used discarded fluid from plasmapheresis treatments for many of our studies because it is a source of large amounts of plasma for studies of permeability activity and for purification of active components. We have surveyed the permeability activity of sera from a wide range of patients with nephrotic syndrome and renal disease. In our initial report, Palb >0.5 was induced by about 30% of FSGS samples tested [45]. In contrast, no increase in permeability was found after incubation with the sera of patients with MCNS or de novo membranous nephropathy after transplantation. We subsequently reported that the sera of 42% (11/26) of children who presented with idiopathic nephrotic syndrome had permeability activity and that Palb did not discriminate between steroid-responsive and steroid-resistant patients [46]. Palb is very high in nearly every patient with collapsing glomerulopathy [47].
Insights from post-transplant recurrence of nephrotic syndrome and FSGS
Therapy of recurrent FSGS includes early plasmapheresis is intended to remove injurious substances(s) [48], [49]. Immunoadsorption using Protein A or polyclonal antibodies to human immunoglobulins has also been used in primary or recurrent FSGS [50], [51]. Many patients have a prompt decrease in proteinuria after therapy. Possible alternative interpretations of the observed benefit of plasmapheresis and immunoadsorption include the addition of a salutary substance or immunomodulation. High doses of calcineurin inhibitors, such as cyclosporine A or tacrolimus, may also improve proteinuria and stabilize renal function in FSGS [52], [53]. Cyclosporine A does not, however, decrease circulating permeability activity in patients with FSGS (2). These agents likely have multiple targets. Cyclosporine A prevents the increase in Palb after incubation with FSGS serum [26], [54] and it also stabilizes the podocyte actin cytoskeleton by blocking the effect of, calcineurin, a serine/threonine kinase, on synaptopodin [55]. We speculate that long-term remissions following relatively short courses of plasmapheresis may be related to the protective effects of calcineurin inhibitors or other agents. It is also possible that susceptibility to injurious agents is enhanced by ischemic or immunological injury at the time of transplant, and that recovery from these acute insults confers some degree of resistance.
Pretransplant plasmapheresis appears to prevent or delay recurrence in patients at high risk for relapse [56]. This response adds support to the concept of removal of an injurious substance. Unfortunately, there is still a high risk for recurrence and repeated plasmapheresis treatments or other therapies may be required to prevent proteinuria and to prolong allograft function [52], [57]. Encouragement regarding potential the efficacy of pretransplant immunotherapy comes from a trial in which transplant patients received hematopoietic donor cells following a nonablative preconditioning regimen. Early recurrence of FSGS was significantly reduced, although only minimal donor-derived engraftment occurred [58]. The precise mechanism for this protection is not well defined, but may include suppression of synthesis of a permeability factor by the conditioning regimen or by the chimeric state. Recent studies have documented a mechanism by which rituximab, a monoclonal antibody to the B cell surface marker CD20 that depletes B cells, may result in remission of FSGS [59], [60]. This agent has traditionally been exclusively thought of as an immunomodulator. It has now been shown to interact with sphingomyelinase of the podocyte and prevents cellular reactions to FSGS serum [8].
The FSGS factor
Approaches to the identification of the ŌĆ£FSGS factorŌĆØ
Initial studies to document the presence of a plasma permeability factor were carried out by infusing plasma into rats. Initial success [61] was followed by discouraging variability in responses to plasma for different individuals. We have studied the function of glomeruli after isolation from the renal cortex [21]. Others have identified candidate proteins based on known modifiers of glomerular function and have measured these in patients with FSGS with or without recurrence [37]. Additional groups have applied proteomic techniques to attempt to determine differences between patient and normal plasma composition [62]. In each case, patient control groups, including those with nephrotic syndrome due to other etiologies and those with uremia, must be studied in order to interpret findings. A functional assay or animal model is also required for the evaluation of the effects of candidate proteins. Injection into animals has proven cumbersome and time consuming. Assays based on the responses of cultured podocytes depend on changes in the configuration of the actin cytoskeleton, cell morphology, and protein expression [55], [63], or measurement of the diffusional permeability of monolayers on a permeable support [64], [65]. Of note, cultured podocytes do not form a confluent monolayer and do not develop slit-pore junctions. Further investigation will be needed before these models are validated as means of characterizing agents that alter glomerular permeability.
In contrast, we have been able to study glomerular barrier function independent of the systemic humoral environment or hemodynamic forces [21]. In addition, more chronic responses can be studied using glomeruli from animal models of disease or after in vivo manipulations [66], [67], [68]. Glomerular cells in isolated glomeruli maintain their complex structure, interaction with other glomerular cells and extracellular matrix, and synthetic capacity [21], [69], and features that are recognized as crucial to integrity of the permeability barrier. Like other technically demanding assays, appropriate effort and time are required to standardize the assay. Nonetheless, several other laboratories have successfully established this technique [66], [70]. The results of studies of isolated glomeruli are complementary to other assays of glomerular and podocyte function.
We have used the Palb assay to verify activity during the sequential purification of proteins from FSGS plasma. We have shown that the active substance in FSGS serum or plasma is a small protein with one or more glycation sites. It has high affinity for Protein A. It has a hydrophobic domain, as evidenced by affinity in a hydrophobic interaction column. During initial fractionation studies, we employed ammonium sulfate precipitation and found that Palb activity was present in a fraction with an apparent molecular weight of about 50 kDa. All the activity of the original specimen was retained in this fraction. Palb activity/mg protein was enriched by more than 10,000-fold compared to the activity of the initial plasma specimen [22], [71]. More recent studies (unpublished data) using affinity chromatography and fractionation by membrane sieving indicate that the Palb activity is present in a fraction with an apparent molecular weight of 30 kDa or less. With progressive purification, it is likely that noncovalently or otherwise loosely-associated proteins are removed, accounting for changes in the apparent molecular size of the active substance. Recently, we have used galactose affinity purification to enrich specimens and have employed mass spectrometry to identify the proteins in the active fraction [72]. Galactose affinity chromatography has allowed us to achieve a high degree of enrichment of activity in a single step.
We have subjected enriched material to mass spectrometry and have identified CLC-1 in the active fraction [36]. CLC-1, a member of the IL-6 family, is the only cytokine present in patient material and not detected in normal plasma. Further studies suggest that CLC-1 may be the permeability factor in recurrent FSGS. It is present in active patient plasma, it mimics the effects of FSGS plasma on Palb, and it decreases nephrin expression by glomeruli and cultured podocytes. Strikingly, a monoclonal antibody to CLC-1 blocks the Palb effect of active FSGS sera. The concentration of CLC-1 in the circulation of patients with recurrent FSGS may be up to 100 times higher than in normal subjects. Studies to confirm and expand these observations are ongoing.
Functional properties of the FSGS permeability factor and clinical associations
The permeability factor in FSGS plasma not only has a strong affinity for galactose, as noted above, but its activity in the Palb assay is blocked by galactose in concentrations of 10ŌĆō12 M [72]. Activity is also blocked by normal serum or plasma [23]. Palb activity is higher in patients with severe and rapidly progressive disease than in those with slower progression [11]. Interestingly, activity has been also been found during post-transplant recurrence in some patients with genetic mutations in podocyte proteins [11], [73]. We can only speculate about the relationship between podocyte mutations and circulating permeability factors. It may be that the co-occurrence is happenstance, i.e. two relatively common conditions coexist, whereas it may be that podocyte abnormalities predispose to injury by circulating substance(s) and result in severe and recurrent renal disease. Further study will be required to understand the relationship between these mechanisms of glomerular injury.
Implications for future therapy
We have identified a number of experimental agents that inhibit Palb activity. These include normal plasma [23], eicosanoids including 20-HETE [24] or 8,9-EET [25], COX inhibitors [74], oxygen radical scavengers [75], certain glycosides of Tripterygium wilfordii
[76] and cyclosporine A [54]. Each of these prevents the increase in Palb induced by FSGS plasma. In the future, these observations may lead to specific therapeutic interventions in FSGS.
Standard therapy and potential new therapies
Steroid therapy is a reasonable first-line therapy and is likely to be effective in the early stages of disease, especially in children [10]. Cyclosporine A or Mycophenolate mofetil (MMF) may induce remissions in additional patients, as shown in a recent trial in children and young adults [77]. The relative effectiveness may be approximately the same. Control of blood pressure, the use of angiotensin converting enzyme inhibitors and/or angiotensin receptor or aldosterone blockers and control of lipid levels with statins or fibrates, appear to provide useful adjunctive measures by which to decrease proteinuria and slow progression. New agents that block the effects of renin, vasopressin or endothelin may be useful in limiting podocyte activation as well as in decreasing proteinuria and controlling blood pressure, but these agents have not been formally tested.
New therapies for patients with steroid-resistant or -recurrent nephrotic syndrome/FSGS are much needed and multicenter trials will be required. The design of these trials will depend on understanding the mechanisms of glomerular injury and/or the progression of renal damage. A multicenter Phase I trial of rosiglitazone and of adalimumab has been completed [78], [79], [80]. The related reports define the pharmacokinetics of these two agents. The salutary responses in some patients and the low incidence of adverse effects support their potential clinical utility. Recruitment for a Phase II trial (IND#103,147) to compare standard conservative therapy (lisinopril, losartan and atorvastatin) versus standard therapy plus adalimumab or standard therapy plus galactose is complete and the study is ongoing. The rationale for using adalimumab is that its antifibrotic activity may slow down or prevent the progression of renal disease independent of its effects on proteinuria. The potential efficacy of galactose therapy is supported by a case report of remission of nephrotic syndrome after oral galactose therapy [81] and complete loss of FSGS permeability activity in a single patient with post-transplant recurrence [72]. Other agents under consideration or in small pilot trials include anti-TGF Transforming growth factor beta (TGF-╬▓) and rituximab [8], [82].
Summary
There is strong evidence that circulating factors may play a role in both MCNS and FSGS in native kidneys, and in recurrent disease in renal allografts. The precise nature of these factors and the mechanisms by which they cause renal injury are the subjects of intense investigation.
At the present, therapy is largely empiric and side effects are significant. In the near future, the mechanisms by which the permeability factors act to cause disease may be understood and rational, targeted therapy with improved efficacy and diminished toxicity may become available. Some therapeutic strategies are depicted in Fig. 2. We strongly believe that ongoing studies to define the nature of circulating injurious factors and the mechanisms by which they alter the permeability barrier and result in kidney injury must continue. Multicenter cooperative studies will be required to test the efficacy of novel therapies. Broad participation in clinical trials will be essential for treating patients with FSGS in native kidneys in order to prevent progression, and prior to and following transplantation to permit long-term graft function and return to health.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









