A unified pathogenesis for kidney diseases, including genetic diseases and cancers, by the protein-homeostasis-system hypothesis
Article information
Abstract
Every cell of an organism is separated and protected by a cell membrane. It is proposed that harmony between intercellular communication and the health of an organism is controlled by a system, designated the protein-homeostasis-system (PHS). Kidneys consist of a variety of types of renal cells, each with its own characteristic cell-receptor interactions and producing characteristic proteins. A functional union of these renal cells can be determined by various renal function tests, and harmonious intercellular communication is essential for the healthy state of the host. Injury to a kind of renal cells can impair renal function and induce an imbalance in total body health. Every acute or chronic renal disease has unknown etiologic substances that are responsible for renal cell injury at the molecular level. The immune/repair system of the host should control the etiologic substances acting against renal cells; if this system fails, the disease progresses to end stage renal disease. Each renal disease has its characteristic pathologic lesions where immune cells and immune proteins, such as immunoglobulins and complements, are infiltrated. These immune cells and immune proteins may control the etiologic substances involved in renal pathologic lesions. Also, genetic renal diseases and cancers may originate from a protein deficiency or malfunctioning protein under the PHS. A unified pathogenesis for renal diseases, including acute glomerulonephritis, idiopathic nephrotic syndrome, immunoglobulin A nephropathy, genetic renal diseases such as Alport syndrome, and malignancies such as Wilms tumor and renal cell carcinoma, is proposed using the PHS hypothesis.
Introduction
Kidneys are constructed of a variety of different renal cell types. Each renal cell has its own cell-fate and characteristic cell-receptors and produces characteristic proteins that cannot be produced by other renal cells. Injury of a kind of renal cells impairs total renal function and induces an imbalance in total body health. Although the etiology or precise pathogenesis of most of renal diseases remains unknown, it is clear that there are etiologic substances associated with all acute or chronic kidney diseases. Each renal disease has its characteristic pathologic lesions. In renal biopsy findings, various immune cells and immune proteins, including immunoglobulins and complement (C) proteins, are commonly seen. These immune cells and immune proteins originate in the host and are observed not only in renal disease lesions, but also in all pathologic lesions of nearly all human diseases. Therefore, it is believed that immune cells and immune proteins may perform the same function in all pathologic lesions associated with the diseases. It has been proposed that the immune system of the host controls the etiologic substances harmful to host organ cells [1]. The author has proposed a unified model of immunopathogenesis for infectious diseases and infection-related immune diseases, including Mycoplasma pneumoniae pneumonia, influenza pneumonia, acute respiratory distress syndrome, and Kawasaki disease, based on the protein-homeostasis-system (PHS) hypothesis [1–5]. In this paper, a common mode of pathogenesis for kidney diseases from acute glomerulonephritis (AGN) to renal cell carcinoma based on the PHS hypothesis is proposed.
The protein-homeostasis-system hypothesis
Multicellular organisms are constructed of numerous kinds of cells whose main constituents are proteins. Furthermore, all biological activities, including embryonic development, biochemical reactions, physiological phenomena and pathologic processes, are performed by proteins that are encoded in genomic deoxyribonucleic acids (DNAs). Therefore, intracellular and systemic protein homeostasis should be maintained for the health of the host. Because every cell of an organism is separated and protected by a cell membrane, communication across cells is required. The intercellular communications are performed by mainly proteins such as cytokines for maintenance of the healthy state of an organism. Thus, the healthy state of the organisms may be controlled by an interacting protein network, designated the PHS [1].
There are etiologic substances involved in all human diseases. The substances may have an affinity to certain target organ cells and they bind and signal the cells, since the eventual symptoms and signs of all diseases result from cell injury at a molecular level. The etiologic substances may be of variable size and possess different biochemical characteristics. They can be classified largely as substances originating from exogenous sources, such as pathogens and natural toxins from animals, plants, and environment factors, or as substances originating from the host cells, such as damage (or danger)-associated molecular patterns (DAMPs) from injured host cells and proteolytic enzymes and proinflammatory cytokines from activated immune cells. The etiologic substances are also classified into two parts: the protein substances and the non-protein substances. The protein substances, including previously named pathogenic proteins (PPs), may have a variety of sizes, similar to monoamines, small peptides (3–10 amino acid residues) such as neuropeptides and peptide hormones, larger peptides (12–30 amino acid residues) that can attach to T cell receptors (TCR), small proteins, and larger protein complexes. The non-protein substances that can be harmful to host cells are also variably sized, including elements such as pure oxygen and carbon monoxide, drugs (chemicals), natural biochemicals such as vitamins and fatty acids, lipopolysaccharides, DNAs and ribonucleic acids (RNAs), and other large complexes (Fig. 1). It is proposed that the immune/repair system is a part of the PHS of the host, and that it controls these substances based on the size and biochemical property of the substances. Briefly, the adaptive immune system controls protein substances according to their size and biochemical characteristics through the recombination of B cell receptor and TCR genes; B cells control medium-sized proteins via the production of antibodies, while T cells control peptides, sized 12–30 amino acid residues, which are controlled by the TCR-associated immune response but cannot induce antibodies. Additionally, smaller peptides (< 12 amino acid residues) may be controlled by a defence system other than the adaptive immune cells in the PHS. The innate immune system cells and immune proteins, including natural antibodies, complements and possibly immune peptides, may control variably sized non-protein substances. Large complex materials, such as whole pathogenic agents, including bacteria and viruses, and large pieces of destroyed cells, such as apoptotic bodies or necrotic debris, are eliminated by neutrophils and phagocytic monocytes through phagocytosis. Furthermore, small non-protein substances, such as pathogen-associated molecular patterns (PAMPs) and DAMPs, are controlled by innate immune cells through pattern recognition receptors, including toll-like receptors and intra-cellular sensors, and immune proteins including natural antibodies (Fig. 1).
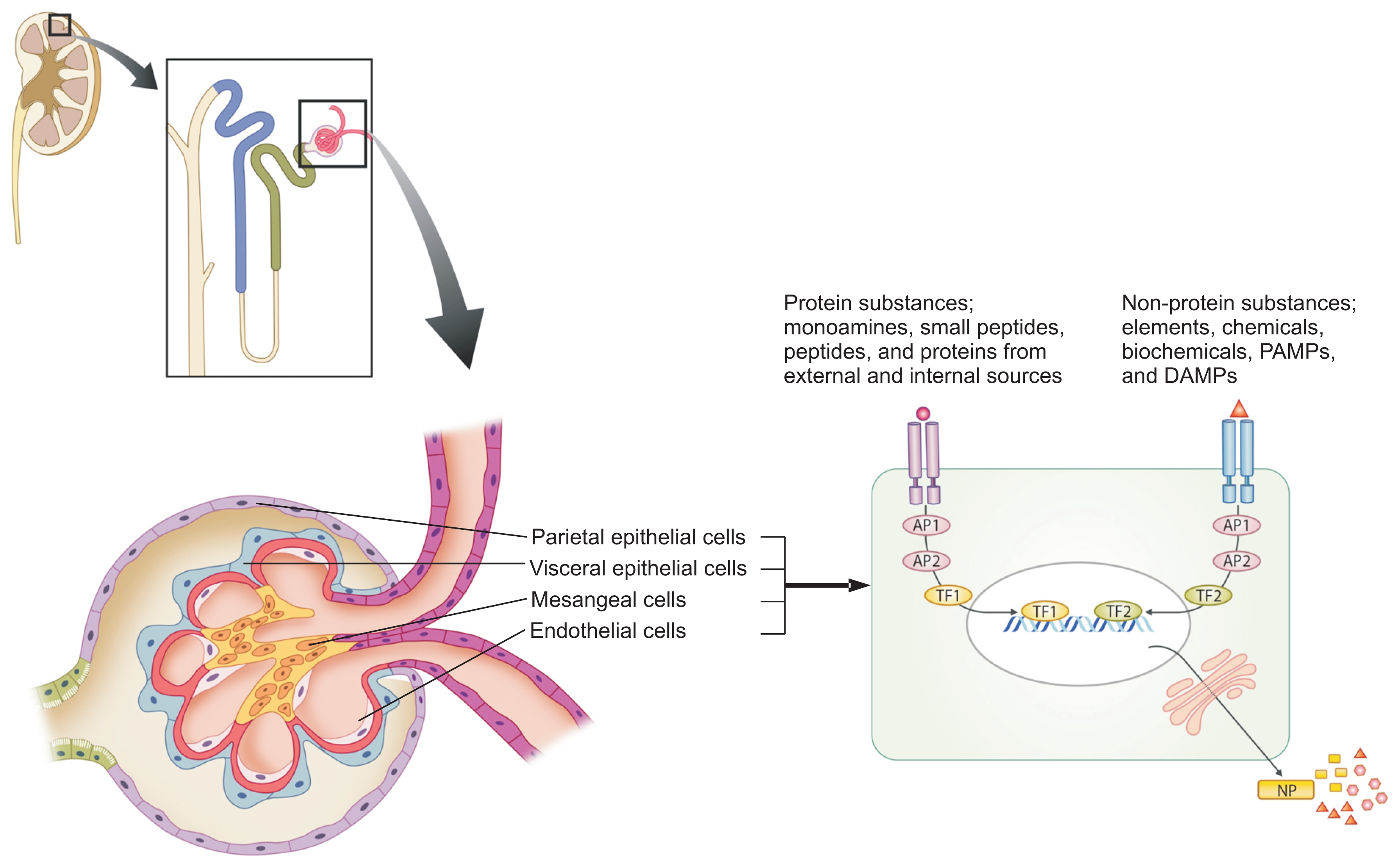
Immunopathogenesis of renal diseases
All biological phenomena in organisms are controlled by a network termed the protein-homeostasis-system (PHS), and the immune system is one aspect of the PHS of the host. Each renal cell has its own receptors for intercellular communication. There are toxic or etiologic substances that have an affinity to renal cells and induce renal cell injury in every renal disease. These substances have various sizes and biochemical characteristics, and they can largely be classified as protein substances and non-protein substances. The protein substances include monoamines, neuropeptides, peptides (12–30 amino acids) and proteins, and each of them originates from external sources such as pathogens and natural toxins or from internal sources such as injured host cells or activated immune cells including heat shock proteins and proinfliammatory cytokines. The non-protein substances include elements (pure oxygen, carbon monoxide, and others), chemicals and biochemicals (drugs, vitamins, fatty acids, and others), PAMPs (lipopolysaccharides, pathogen DNAs and RNAs, and others), and DAMPs from the host cells. Each immune cell and immune protein may recognize and act on these substances, based on the size and biochemical characteristics of the substances; innate immune system cells control non-protein substances, and adaptive immune cells control protein substances including pathogenic proteins and pathogenic peptides. The severity, chronicity or prognosis of renal diseases depends on the amount of etiologic substances with corresponding immune reactions, the duration of the appearance of specific immune cells, or the existence of specific immune cell repertoires that control the substances.
AP, adaptor protein; DAMPs, damage-associated molecular pattern; NP, new protein; PAMPs, pathogen-associated molecular patterns; TF, transcriptional factor.
It is also proposed that the PHS may control the situation associated with a defective protein or protein deficiency [1,3]. Given that a protein defect can induce an early fatal outcome in congenital nephrotic syndrome and severe immunodeficiencies, genetic diseases and cancers may also originate from a protein-deficiency or a malfunctioning protein under the PHS.
Acute glomerulonephritis
The prototype of AGN is acute poststreptococcal glomerulonephritis (APSGN), a representative immune-mediated disease which occurs after a known bacterial infection, such as group A β-haemolytic streptococci (GAS) [6]. Acute onset of the disease occurs after the initial symptoms of the GAS infection, such as pharyngitis or pyoderma, have subsided and at that time, hypergam-maglobulinaemia, i.e., elevated serum immunoglobulin G (IgG), IgA and IgM, and hypocomplementaemia (low C3) are already noted [7]. Thus, it is possible that, during the recovery stage from the initial GAS infection, the substances that induce renal inflammation are produced and reach glomeruli through systemic circulation. The incubation period ranges from 1 to 4 weeks from the initial symptoms and signs of GAS infection with individual variations. Some patients with APSGN or acute rheumatic fever have no prodromal symptoms or signs of initial GAS infection, manifesting as an asymptomatic infection. In addition, early systemic antibiotic therapy for initial GAS infection cannot prevent the development of APSGN and does not affect the natural course of APSGN [8], suggesting that the substances may not originate from the pathogens. The proposed substances include the structural antigens of GAS, such as M proteins, and secretory nephritogenic proteins, such as nephritis-associated plasmin receptor and streptococcal pyrogenic exotoxin B [9]. Moreover, other pathogens such as group C streptococci, staphylococci, and viruses can induce postinfectious glomerulonephritis [10].
In the early pathologic lesions of APSGN, glomerular deposits of C3 and IgG are seen along capillary loops and often in mesangial areas. Many neutrophils, but rarely lymphocytes, are seen in capillary loops. Serum IgG and C3 levels are not related to the clinical severity during the acute stage of the disease [11], and many patients have no IgG deposits except C3 in renal lesions [12]. It was reported that the IgG level and ASO titre do not change, while C3 and IgM levels significantly increase during the acute stage of APSGN [13]. These findings suggest that the innate immune system, including complements, neutrophils, and natural IgMs, may play an important role in recovery from the early stage of the disease. Most of patients with APSGN in children recover from the disease without complications.
Crescentic glomerulonephritis (CGN) or rapidly progressive glomerulonephritis is a severe form of GN whose characteristic histopathologic finding is crescent formation in the majority of glomeruli [14]. The crescents in Bowman’s space are mainly composed of proliferated parietal epithelial cells, macrophages, and other proteins such as fibrin. These pathologic characteristics are observed in all types of severe renal cell inflammation and subsequent cell injury, regardless of the deposition of immune complexes. Therefore, the substances that induce crescent formation may affect all parietal epithelial cells in both kidneys and may originate from common host cells, such as activated immune cells and/or possibly injured renal cells rather than from various pathogens. In combination with immunosuppression, plasmapheresis (or plasma exchange) has been reported to benefit patients with CGN and other intractable autoimmune diseases [15]. Plasmapheresis targets etiologic substances in plasma for removal, but it may have limited effectiveness on substances readily bound to target cells or continuously produced substances. The prognosis of most forms of CGN is grave, and most of patients with CGN progress to end stage renal disease (ESRD).
After an infection, including GAS infection, a variety of substances that have affinity for different renal cells and they may originate from the pathogens or host cells injured by infectious insult. Although the immunopathogenesis of AGN from APSGN to CGN may be complex, the host immune/repair system eventually controls this renal disease and determines the prognosis of the disease. The prognosis of AGN, from APSGN to CGN, may be dependent on whether the host immune/repair system can control the etiological and other toxic substances produced during the inflammatory processes or cannot.
Idiopathic nephrotic syndrome
Nephrotic syndrome is caused by massive proteinuria (> 40 mg/m2/hour in children) and is characterised by hypoalbuminaemia (< 2.5 g/dL), hyperlipidaemia, generalized oedema, and hypogammaglobulinaemia, i.e., low IgG [16]. Idiopathic nephrotic syndrome is (INS) the most common cause of nephrotic syndrome in children and accounts for 90% of all childhood nephrotic syndrome. Approximately 85% of INS patients are steroid-sensitive. Histopathologically, 3 types of lesions have been described: minimal change, mesangial proliferation, and focal segmental glomerulosclerosis (FSGS). These pathologic lesions are associated with steroid responsiveness and prognosis; the initial response rate is 90% in minimal change nephrotic syndrome (MCNS), but 10% in FSGS, and nearly all steroid-resistant nephrotic syndrome (SRNS) progresses to ESRD. The glomerular filtration barrier consists of fenestrated endothelial cells, glomerular basement membrane (GBM), and highly specialised epithelial cells termed podocytes. The inter-digitating foot processes of podocytes form a slit diaphragm which consists of several structural proteins and performs ultrafiltration of molecules through signalling to the cytoskeleton of the podocyte [17]. Injury of podocytes and/or the GBM barrier induces proteinuria, and the severity of proteinuria and the size of the filtered proteins in urine can be influenced by the severity of GBM injury in renal diseases [18]. INS, including congenital nephrotic syndrome, results in highly selective proteinuria, i.e., few IgG excretion in urine, despite a markedly lower level of serum IgG. In steroid-sensitive nephrotic syndrome, serum albumin and IgG levels show a positive correlation, and albumin and cholesterol levels show a negative correlation [19]. This may be a characteristic of the nephrotic syndrome resulting from podocyte injury only, and other forms of injury to the GBM barrier may induce non-selective proteinuria with IgG excretion. The cause of hypogammaglobulinaemia in MCNS is unknown. Because maintenance of oncotic pressure by high-molecular lipoproteins that cannot pass the GBM pores may be essential in INS, it is proposed that IgGs may move to an extravascular compartment for the intravascular space to larger lipoproteins [20]. A genetic screening test for congenital or familial nephrotic syndrome is now well established. Ten to twenty percentage of patients with SRNS have genetic mutations in genes coding key podocyte proteins; these genes include NPHS1, NPHS2, TRCP6, CD2AP, and ACTN4 [17,21]. These gene products constitute the slit diaphragm or the podocyte cytoskeleton. However, other genes encoding GBM or mitochondrial or transcription factors, such as the Wilms tumour gene (WT1), are associated with SRNS [21].
The etiology of INS is unknown, but children with INS may have had a preceding infection with an unknown pathogen(s) prior to disease-onset. Like APSGN, changes in the epidemiology of INS have occurred in various populations; incidence of INS has been markedly reduced compared to the past in developed countries and may be also decreasing in developing countries in association with economic growth and improvements in public hygiene [22,23]. There may be substances that induce proteinuria in INS, and the target cells of these substances may be epithelial cells (podocytes) in the glomerular filtration barrier. T cell-origin soluble factors have been proposed, because of an absence of immune deposits, transient remission after measles infection which could suppress T-cell function, occurrence of nephrotic syndrome in Hodgkin lymphoma, and the response to immunosuppressive agents such as corticosteroids and cyclosporine A [24]. Other presumed substances for FSGS include cardiotrophin-like cytokine factor 1, soluble urokinase plasminogen activator receptor, and anti-CD40 antibodies [25,26].
In the pathologic lesion of MCNS or early stage of FSGS, there is no immune cell infiltration visible by light microscopy (LM), and no immunoglobulin or complement precipitations are identified by immunofluorescence (IF). Only effacement and fusion of foot processes is seen by electron microscopy (EM) [27]. The etiologic soluble permeability factors may also originate not only from pathogens, including PAMPs, but also from host cells, including DAMPs, from injured cells or activated immune cells. It is postulated in the PHS that the substances that induce these pathologic findings may be small peptides fewer than 12 amino acids that cannot be controlled by T cells and B cells [1]. The small substances may bind to receptors on the podocytes, and the substance-receptor complex signals the nucleus to produce new proteins or induce apoptosis. This event or other substances from the PHS which control these peptides could affect the podocytes or the proteins in the cells. During the natural course of INS, two-thirds of MCND patients experience relapses, especially when they have infections, such as an upper respiratory infection, caused by various pathogens. This finding also suggests that the etiologic substances may be associated with host immune reaction. Possible explanations include that the etiological substances are produced in greater amounts during new infections, or that the control system against these etiologic substances needs more works against new substances produced by the infections. Certain diseases, including acute rotavirus infection and chronic neurodegenerative disorders such as prion diseases and Alzheimer disease, few immune cells and antibodies can be detected in early pathologic lesions [28–30]. It is possible that the etiologic substances causing cell injury in these diseases may also be small peptides controlled by an unknown immune system in the host. It has been reported that immune-suppressants of B cells (rituximab, an anti-B cell agent), as well as those of T cells, were effective in steroid-dependent nephrotic syndrome [31]. Because all immune system cells in the PHS may communicate with each other against any immunological insults caused by external or internal substances [1], blockage of a pathway in the hyperactivated immune reaction may downregulate the events of proteinuria. Some FSGS patients were shown to relapse rapidly after renal transplantation, and this may have been suggested due to circulating soluble factors [25,26]. Thus, the substances that elicit podocyte injury may be continuously produced from extrarenal foci, and if we could identify and remove the foci, the disease could be curable. It is possible that the PHS of children with FSGS cannot control the pathogenic substances that act against podocytes, and that continuous activation of a non-specific control system, including immune cells with cytokines, induces FSGS and results in permanent renal cell injury.
IgA nephropathy
IgA nephropathy is a representative chronic glomerular disease in children and young adults. Diagnosis of the disease is based on renal biopsy; the characteristic finding is a predominance of IgA deposits within the mesangial area in the absence of systemic disease [32]. The clinical manifestation of childhood IgA nephropathy, including duration of the illness, varies from asymptomatic microscopic haematuria to rare severe CGN. The clinical presentation in children is benign in comparison to adults, and 20–30% of children with IgA nephropathy are persistent to adulthood, and a large proportion of them may experience ESRD later in life [33]. Gross hematuria often appears within 1–2 days of the onset of an upper respiratory infection, and a normal serum level of C3 helps to distinguish IgA nephropathy from APSGN.
In pathologic lesions, focal and segmental mesangial proliferation (increased mesangial cells and matrix) are seen in LM findings. Additionally, epithelial cell crescent formation and sclerosis in the glomerulus can occasionally be seen. C3 deposits in the mesangium are often accompanied with IgA deposits in IF findings. Thus, IgA and complements may play roles in controlling the etiologic substances whose target cells may be mesangial cells, although an abnormal structure of IgA has been proposed as the cause of IgA nephropathy [34]. In addition, serum IgA level is elevated in only 15% of pediatric patients, and the levels of IgA not associated with prognosis of the disease as likely in IgG levels in APSGN. The precipitated immunoglobulins in pathologic lesions vary according to renal disease entity. IgG is the main immunoglobulin precipitate along capillary walls in membranous proliferative glomerulonephritis and often in APSGN. In rare renal disease entities such as IgM nephropathy, immunotactoid glomerulopathy and cryoglobulinemic nephropathy, immunoglobulins (IgM and/or IgG) are also found in renal tissues [35,36]. IgG4-positive plasma cells and IgG4 deposition are seen in IgG4-related kidney diseases [37]. The etiologic PPs of IgA nephropathy may have a specific size and biochemical characteristic that may be suitable for control by IgAs, but not by IgGs or IgMs in the PHS hypothesis. IgA infiltration in glomeruli is observed not only in IgA nephropathy, but also in other conditions such as Henoch-Schölein purpura (HSP) nephritis, alcoholic liver cirrhosis, and IgA-dominant postinfectious GN [38–40]. IgA or IgA plasma cell infiltrations are seen in the skin lesions of HSP and in other diseases, including in hepatic cells in alcoholic liver cirrhosis and kidney and other organ cells in fatal Kawasaki disease patients [41]. IgA nephropathy can recur after renal transplantation, and this suggests that the substances that induce IgA infiltration and inflammation in the mesangial areas may originate from extrarenal foci as well as FSGS. Although the renal pathologic findings in IgA nephropathy and HSP nephritis are similar, patients with HSP experience purpura, abdominal pain, arthralgia, and signs of other organ cell involvement, likely due to additional substances that affect extrarenal cells. Taken together, these results indicate that immunoglobulins, including IgA, found in renal tissues may be secondary phenomena derived from systemic immune reactions rather than primary etiologic agents [34]. The etiologic substances may be released from other host cells which are injured by hyper-activated immune cells or other factors and reach mesangial cells via systemic circulation. Tonsillectomy has been recommended as one treatment modality for IgA nephropathy, although prospective, controlled trials are lacking [42]. The effect of this treatment could be explained in part by the elimination of the foci producing the etiologic substances. Although corticosteroids or immune suppressants can reduce proteinuria and improve renal function in patients in the early stage of the disease, it remains unclear if the effects of glucocorticoids and immune-suppressants deter progression to ESRD [32,34]. Immune-modulators may be ineffective in removing the etiologic substances in this chronic GN.
Immunopathogenesis of other renal diseases, including acute pyelonephritis and tubulointerstitial diseases, could be explained by the same way, and there are etiologic substances that have affinity to target renal cells and corresponding immune reactions in each renal disease [43].
Genetic diseases
Because the kidney consists of a variety of renal cells, there are numerous genetic renal diseases that originate from each type of renal cell [44]. A variety of clinical manifestations of a genetic disease may be caused by protein defects, such as a protein deficiency or a malfunctioning protein. Certain genetic renal disease, such as congenital nephrotic syndrome, can be fatal during early infancy, as is the case with severe immunodeficiencies derived from a protein defect on T cells or neutrophils [45,46]. In general, autosomal dominant genetic diseases are caused by structural protein defects, and autosomal recessive diseases are caused by defects in enzymatic proteins. However, certain genetic diseases that have an enzymatic protein defect can induce structural cell injury during the natural course of the illness. In addition to this phenomenon, there are unsolved enigmas in genetic diseases. In familial genetic diseases such as Alport syndrome, the clinical manifestations, including the time of onset of clinical symptoms such as proteinuria, severity of the disease and prognosis, can be different in members of the same family, despite the presence of the same genetic defect. Consequently, the phenotypes expressed may not be identical despite the presence of the same genotype [47]. Additionally, genetic diseases are genetically heterogeneous; similar phenotypes can occur from different genotypes [48]. The most common genetic type in Alport syndrome is an X-linked mutation in the COL4A5 gene encoding the α5 chain of type IV collagen. GBM consists of 3 chains of collagen, the α4, α5, and α6 chains. However, patients with this structural protein defect show no clinical symptoms or LM findings during the first decade of life. Along with progressive proteinuria over time, renal pathologic findings develop including mesangial proliferation, capillary wall thickening, and glomerular sclerosis. In addition, chronic irreversible lesions, such as tubular atrophy, interstitial fibrosis, and lipid-containing tubular or interstitial cells called foam cells, are also seen. Interestingly, despite the structural protein defects, EM findings of GBM are near normal in a portion of patients with any genetic form of Alport syndrome, although most patients show diffuse thinning and thickening, splitting or weaving of the glomerular and tubular basement membranes [49]. These enigmas in Alport syndrome may also be seen in other genetic diseases including autosomal dominant polycystic kidney disease (ADPKD) [50]. In gene knock-out animal studies, the phenotype of single gene knock-out mice varies according to mouse strains, and clinical abnormalities from the gene defect can differ between mice and humans. In addition, mice null for some genes have no observable phenotypic abnormalities compared to controls [51]. How do we explain these enigmas? It is possible that there are certain mechanisms that produce compensatory or alternative proteins in the case of protein defects in vivo. In other words, the PHS in an organism may control the defective proteins or specific protein deficiencies. Thus, patients who can induce proper alternative proteins might have a less severe clinical course and a better prognosis. It is possible that the production of transformed proteins by a mutant gene acts as the PP, or that prolonged activation of these alternative proteins induces substances that are toxic to host cells (Fig. 2). It is unknown whether these alternative proteins act like PPs are expressed during a certain period of life.

Pathogenesis of genetic diseases and cancers under the protein-homeostasis-system (PHS) of the host
A genetic disease is caused by a defective protein or specific protein deficiency (A). In the PHS hypothesis, the PHS of organisms can induce alternative proteins (AP) in various organ cells in need (B). Thus, the patients who produce more proper AP might have a less severe clinical course and a better prognosis in a familial genetic disease. Also, it is possible that prolonged activation of transformed proteins or AP is associated with the substances that are toxic to host cells in natural course of some genetic diseases (C). A cancer cell originates from a gene defect of an intracellular protein which may be essential for cell survival (D). AP are produced within the cells with new gene activation, and these proteins may also be controlled by the intracellular PHS if the cell avoids cell death. During this process in replicating cells or stem cells, serial activation of genes affecting cell-replication cycles, including embryonic transcriptional factors, occurs and an eventual self-reproducing cell can be established (E). In tumor growth in most cancers, the stop signals (proteins) may not function, because of ongoing new proteins that are produced from renewal cancer cells and cannot be controlled by the systemic PHS in the host (F). Whereas normal organogenesis of stem cells such as embryonic development and organ repair is strictly controlled by stop signals (proteins) under the PHS in the host. TP, transformed proteins.
Interestingly, corticosteroid treatment may help delay the progression of some genetic disorders, including Duchenne muscular dystrophy [52]. It is believed that many autoimmune diseases may have genetic traits for disease susceptibility and response to corticosteroids. These findings suggest that certain non-specific, alternative proteins present in genetic diseases could be harmful to host cells, and corticosteroids could alleviate the symptoms and signs in some genetic diseases, as shown in most other diseases, including infectious, allergic, and autoimmune diseases.
Cancers
It is believed that all cells present in our bodies are continuously regenerating, and old cells are replaced to renew tissues. However, the mechanisms and the sites of renewal in specific organs during steady-state are unknown.
There are many kinds of malignancies of renal cell origin. In this review, Wilms tumor and renal cell carcinoma are simply discussed. Wilms tumor, also known as nephroblastoma, is an embryonal-origin malignancy and is the most common primary malignant renal tumor of childhood [53]. It is proposed that these are benign foci of embryonal kidney cells that persist abnormally into post-natal life in new-born kidneys, and genetic factors may predispose an individual to these nephrogenic rests [54]. Also, it is proposed that tumorigenesis in Wilms tumor is associated with embryonal cells in the nephrogenic rests. These cell clusters have a genetic defect, including Wilms tumor gene (WT1), which may sustain additional mutations and transform into tumour cells [55]. In the PHS hypothesis, a cancer cell also originates from a gene defect in an intracellular protein which may be essential for cell survival. Alternative proteins are produced within the cells with new gene activation, and these proteins may also be controlled by the intracellular PHS, if the cell avoids cell death. During this process in replicating cells, serial activation of genes affecting cell-replication cycles, including transcriptional factors, occurs, and an eventual self-reproducing cell can be established. Currently, inducible stem cells as self-replicating cells can be obtained from artificial activation of only 4 transcriptional factors affecting early genes of the embryonic stage [56]. Thus, the alternative-protein theory can be applied to the oncogenesis of renal cell carcinoma and other organ cancers (Fig. 2).
The incidence of renal cell carcinoma, as well as of most cancers, increases as individuals become older [57]. It is known that somatic cells have limitations in the number of cell divisions that they can undergo together with telomere shortening. Therefore, “old” somatic cells that have undergone many division cycles may be fragile enough to sustain a gene mutation and subsequently become cancer cells [58]. Sequential genetic defects in precancerous cells participating in lesion repair, including the activation of oncogenes and/or suppression of tumour suppressor genes, have been suggested as required factors for the establishment of cancers. However, this process is not universal within a given cancer, and it takes several years to decades, indicating that not all of these genes are needed for cancer development. Renal cell carcinoma is associated with genetic defects in various genes, including loss of function of the Von Hippel-Lindau (VHL) tumour suppressor gene, located on chromosome 3p; however, these defects do not appear in all renal cell carcinoma and can appear in other cancers [59].
Some cancers are associated with chronic infections including hepatocellular carcinoma and cervical carcinoma [60,61]. Some malignancies are associated with genetic diseases, such as familial adenomatous polyposis in colorectal cancers or WAGR syndrome and Denys-Drash syndrome that predispose to Wilms tumor [55,62]. It is possible that various tissue cells which are affected with chronic infections or genetic defects may have a short turn-over time of replication with subsequent fragility in a genetic variation, although chronic infections may induce epigenetic changes such as gene methylation and activation of microRNAs in the infected cells. Chronic stimulation by unknown substances may induce persistent activation of the corresponding immune cells, and one of the immune cells or progenitors in the excessive proliferation state may transform into a cancer cell. In hematologic malignancies, acute blast transformation has been observed in chronic lymphocytic leukaemia, chronic myelocytic leukaemia, multiple myeloma, and idiopathic hypereosinophilic syndrome may fall in this category [63,64].
Wilms tumor or renal cell carcinoma is genetically heterogeneous, as is the case with other organ cancers. Genetic information may change continuously as cancer cells undergo further divisions because an initial gene defect may induce continuous alternative-protein production. Thus, the extent of genetic variation in cancer cells may be associated with the number of cancer cell proliferation cycles. Reciprocal translocations of chromosomes have also been observed in some patients with renal cell carcinoma as well as in hematologic malignancies (Philadelphia chromosomes) [65,66]. The resulting oncogene formed from two genes (gene fusion), produces a new protein which may not exist in normal cells, but this protein may be needed in cancer cells; however, such proteins should also be controlled by the intracellular PHS. As more cancer cells grow and accumulate genetic variation, increased gene activation may be needed to maintain protein homeostasis within the cells. Thus, it is natural that malignancies with immature histologic findings, such as anaplasia or gene translocation, may have a poor prognosis because of an increase in new proteins for the PHS (Fig. 2).
It has been postulated that solid cancers originate from stem cells, based on the fact that some cancer cells express stem cell markers of embryonic origin. There are various tissue stem cells in vivo, including bone marrow stem cells, which are involved in tissue regeneration and repair after injury of organ cells [67]. Embryonic stem cells and progenitor cells in organogenesis and tissue-origin stem cells for the repair of an organ construct an organ structure with auxiliary tissue cells and immune cells and stop working when the structure is completed. The stop signals may be influenced by various factors, including cell-to-cell interactions in the microenvironment and a communicating network of proteins systemically controlled by the PHS. Unfortunately, the stop signals (proteins) may not function in most cancers, possibly due to numerous new proteins from the cancer cells that cannot be controlled by the systemic PHS in the host. However, remission of cancers can occur spontaneously on rare occasions and occurs more commonly with early anticancer treatment before multiple replication cycles of the cancer cells have occurred under the control of the PHS which contains immune cells, including natural killer (NK) cells and cytotoxic T cells (Fig. 2).
Today, targeted and personalized treatment for organ-specific cancers has become a main focus of research [68,69]. The growth of cancer cells is influenced by a protein network required for cell-to-cell interactions; thus, blockage of a protein which is involved in the pathways relating to the cell replication cycle or critical intercellular communication can stop cancer cell growth. Cell-cell interactions are performed through signal mediators (proteins) and their own receptors on cancer cells. Identifying the receptors playing a critical role in cancer cell survival, such as inducing apoptosis, is a first step in anti-cancer drug development and is being extensively investigated in renal cell carcinoma as well as in other cancers [70].
In summary, cancers are caused by a breakdown of the PHS within a cell. A protein defect in the replicating organ cells or in the tissue stem cells during organ cell repair may induce sequential reactivation of early embryonic genes or activation of genes required for the cell replication-cycle during continuous production of alternative proteins and controlling proteins for these proteins, and it may be responsible for the establishment of a cancer cell. After the establishment of a cancer cell, cancer growth depends on the proteins present in the microenvironment of the local lesions; however, these proteins should be controlled by the systemic PHS on an individual basis.
Conclusion
All biological activities of an organism, including embryonic development, growth, aging, and immunological responses to diseases, may be carried out by proteins in an order, i.e., the PHS. Therefore, the PHS in organisms may recognize self-proteins, eliminate the proteins harmful to host cells, supply proteins when deficiencies occur, and maintain protein homeostasis in order to establish a healthy state. There are many small protein derivatives (monoamines and peptides) which do not readily incite antibody production, and some of these play significant roles on the homeostasis of an organism. Given that circulating immune cells and immune proteins, including antibodies, complements, cytokines and possibly immune peptides, are the only effectors for the prevention of tissue cell injury and tissue cell repair in the host, it is reasonable that immune cells and immune proteins recognize and act against the size and biochemical properties of substances including PPs and peptides. The PHS hypothesis is very simple, but it could unify the pathogenesis of kidney diseases, including genetic diseases and cancers, as well as the mode of mechanisms in development, growth, and activity of an organism.
Acknowledgments
The author thanks Drs. Hong Su-Ah, Hwang Hyeon-Seok, Seo Jin-Soon, Rhim Jung-Woo, Park Seok-Young and others for review and advice on the manuscript.
Notes
Conflicts of interest
The author has no conflicts of interest to declare.