


Introduction
Syndrome of inappropriate antidiuretic hormone secretion (SIADH) is a disorder that is characterized by hyponatremia and impaired water excretion. Many other disorders of the central nervous system (CNS) have been reported to be associated with SIADH. However, association of the SIADH with neuromyelitis optica (NMO) is very rare. NMO is an inflammatory demyelinating disease of the CNS characterized by severe optic neuritis and longitudinally extensive spinal cord lesions. NMO spectrum disorder (NMOSD) has recently been proposed to refer to the spectrum of aquaporin-4 antibody (AQP4-Ab) related diseases, including definite NMO. It is comprised of several conditions, which include both AQP4-IgG antibody and one of the index events of the disease (recurrent or bilateral optic neuritis and longitudinally extensive transverse myelitis [LETM]). The AQP4-Ab has been described as a relatively sensitive and specific marker for NMO. However, some patients remain seronegative despite repetitive testing. We reported a case of a patient with seronegative NMOSD presenting with hyponatremia.
Case report
A 37-year-old female was admitted to our hospital in May 2015 due to nausea and vomiting, which had lasted for one month. Before she came to our hospital, she was admitted to another hospital and had already undergone esophagogastroduodenoscopy with no specific findings. At the time of admission, she was also complaining of dizziness, fatigue and hiccupping. Her vital signs and neurological examination results were within normal limits. Her tongue was not dehydrated, skin turgor was normal, and she had no pitting edema.
However, the patient was found to have hyponatremia. Her initial serum sodium level was 129 mEq/L. The level of other electrolytes were potassium 4.0 mEq/L, chloride 93 mEq/L, uric acid 0.9 mg/dL, blood urea nitrogen 9 mg/dL, and creatinine 0.6 mg/dL. Her serum osmolality and urine osmolality were 253 and 595 mOsm/kg, respectively. The levels of urine sodium and urine potassium were 195 and 24 mmol/L, respectively. Her thyroid function test was within a normal range.
The patient’s plasma adrenocorticotropic hormone (ACTH) level was 28.5 ng/mL, and we performed rapid ACTH stimulation test, which showed a normal response. In this patient, there was no evidence of other organ dysfunction. The patient was diagnosed with SIADH after exclusion of other causes. Moreover, she did not show any evidence of dehydration, and had normal blood and plasma volume. This sign excluded a diagnosis of cerebral salt wasting (CSW) syndrome.
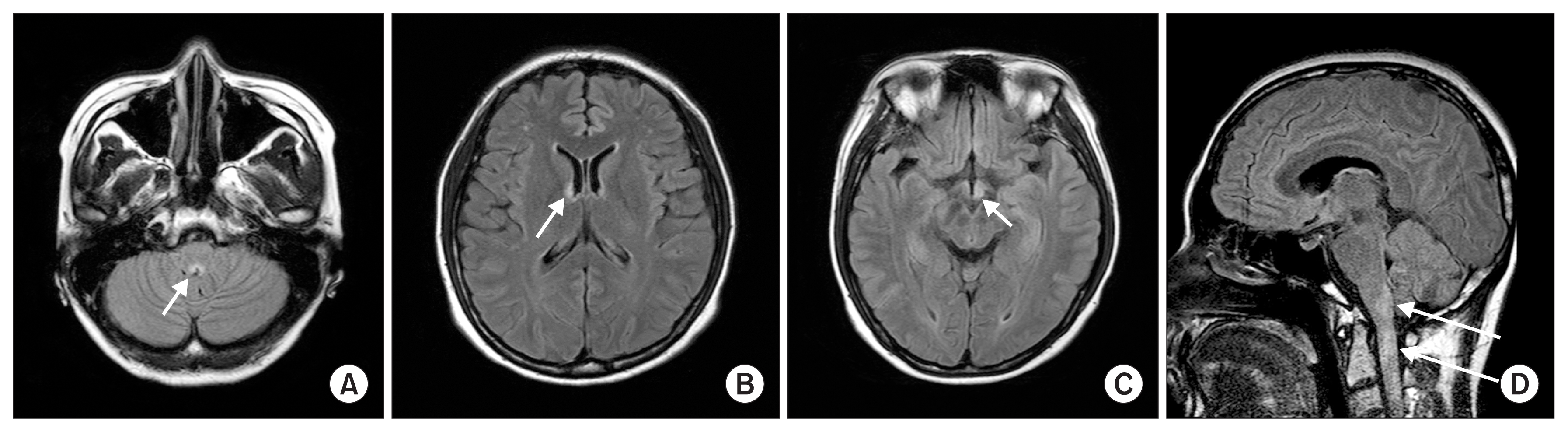
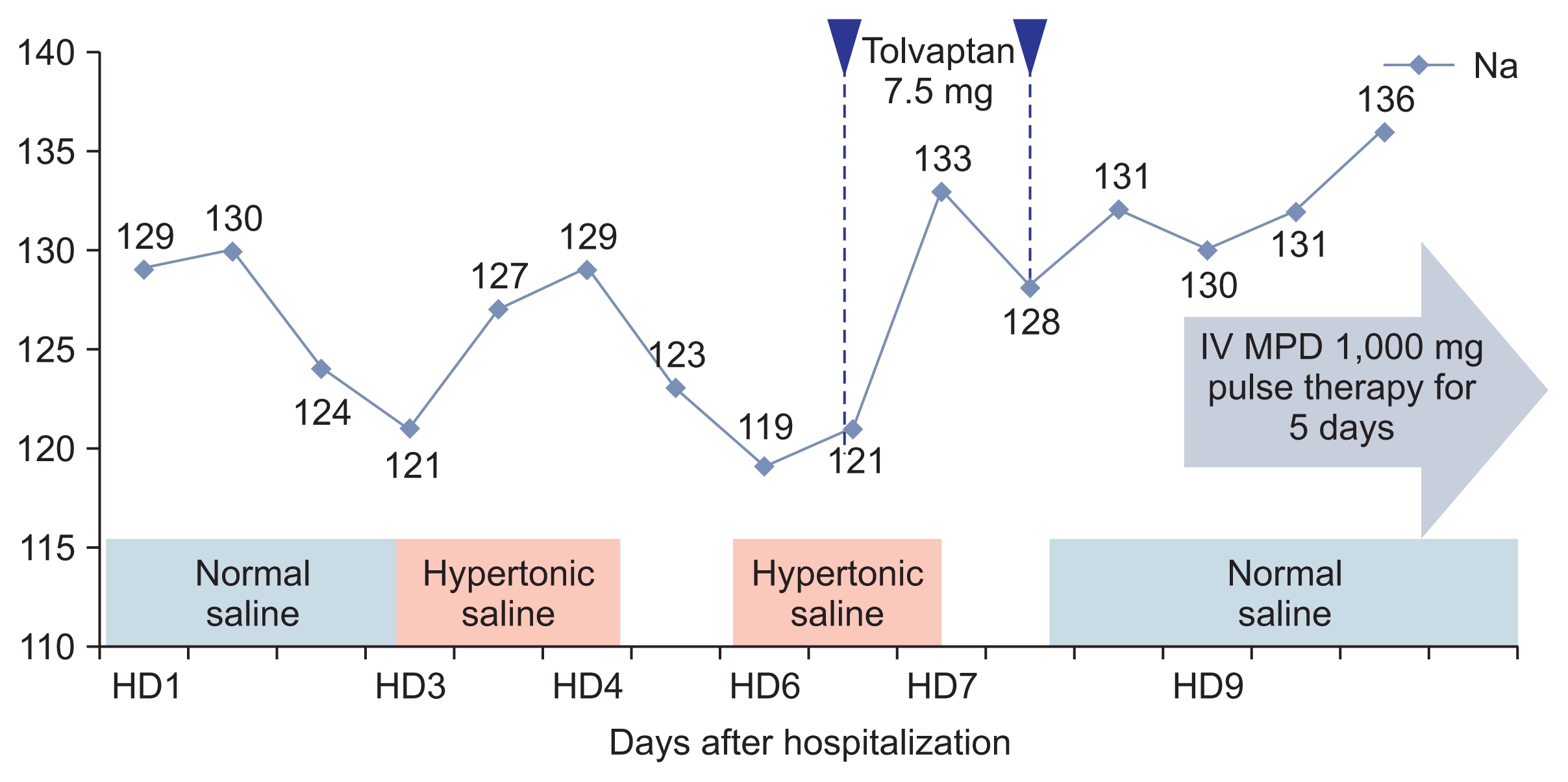
Serum sodium level was decreased from 129 to 121 mEq/L during two days in our case. Correction of serum sodium concentration was achieved by hypertonic saline (3.0% NaCl, intravenously). However, after discontinuation of hypertonic saline, serum sodium level decreased again to 119 mEq/L. Even tolvaptan, a selective vasopressin receptor 2 antagonist, did not resolve the hyponatremia, and at that time the patient complained of hoarseness, diplopia, and marked progression of dizziness. Follow-up neurological examination revealed right ptosis, spontaneous nystagmus, miosis, and gait ataxia, suggesting multiple brain stem lesions. Cerebral T2-weighted magnetic resonance imaging (MRI) showed a multifocal high signal intensity at the lower medulla oblongata, hypothalamus, optic chiasm, subcortical white matter, and upper cervical spinal cord (Fig. 1). Cerebrospinal fluid (CSF) analysis shows CSF pleocytosis (30 cells/mm3 white blood cells count) with lymphocytic predominance (97%), slightly elevated protein (50.6 mg/dL) and normal glucose (88 mg/dL) levels. The serum anti-AQP4 antibody showed borderline levels. After 4 months, anti-AQP4 antibody undetectable. Other serum antibodies associated with autoimmune diseases were all negative. She also did not show any clinical features of vasculitis. Though there was no clinical history of optic neuritis, visual evoked potential showed delayed P100 latency in both eyes. Thus, the patient was diagnosed with NMOSD due to extensive myelitis and a typical brain MRI lesion. The patient was successfully treated with high-dose methylprednisolone (1,000 mg methylprednisolone for 5 days), followed by a gradually tapering course of prednisolone. After high-dose steroid treatment, her neurological symptoms gradually improved and her CSF pleocytosis and serum sodium level became normal. Her clinical course is shown in Fig. 2.
Discussion
SIADH is the most frequent cause of hyponatremia [1]. In this case, the patient showed normal renal and adrenal function, and there were no abnormal findings that could cause hyponatremia. She was diagnosed with SIADH by excluding other causes, and SIADH occurred simultaneously with exacerbation of neurological symptoms. Hyponatremia is a common electrolyte disorder in the CNS and distinguishing between CSW and SIADH in clinical practice can be difficult [2]. However, a normal extracellular fluid volume supports a diagnosis of SIADH rather than CSW. Moreover, this diagnosis is supported by low levels of uric acid and normal plasma albumin concentration and hematocrit.
Several studies have reported cases of demyelinating CNS disorder that presented with SIADH. In particular, the combination of multiple sclerosis and SIADH has been often reported previously [3,4]. However, SIADH rarely occurs in NMO. A literature review revealed that several cases of SIADH have been described in NMO. Lotze et al [5] reported the case of a 15-year-old girl with AQP4-Ab-positive NMO who had symptomatic hyponatremia caused by SIADH attributed to disease involvement of the bilateral hypothalamus on brain MRI. You et al [6] reported on a 56-year-old woman with AQP4Ab-positive NMO who was diagnosed with SIADH. Her brain MRI showed no hypothalamic lesions, but it was speculated that there may have been damage to the tiny connective fibers of the hypothalamus that could not be detected by MRI. Nakajima et al [7] reported the case of a patient with NMOSD who showed SIADH associated with a unilateral hypothalamic lesion as an initial manifestation. In those three cases, unlike in our case, the serum anti-APQ4 antibody test was positive, but all were treated with intravenous methylprednisolone, as in our case, followed by oral steroid taper with a resolution of neurological symptoms and SIADH.
NMO and its limited forms, known as NMOSD, are inflammatory, demyelinating syndromes of the CNS that are characterized by severe attacks of optic neuritis and longitudinal extensive transverse myelitis. NMOSD is associated with the NMO-IgG biomarker, which targets the AQP4 water channel on astrocytes at the interface between the CNS parenchyma and fluid compartments [8,9]. AQP4 has been more highly expressed in the hypothalamic and periventricular areas of the brain. Thus, SIADH may have been caused by anti-AQP4 antibody-mediated inflammation in the hypothalamus. In our case, intractable nausea/vomiting and hiccupping were the patient’s first symptoms. Iorio et al [10] reported that 14% of 70 AQP4Ab-positive patients had initial presenting symptoms of NMO, such as intractable nausea, vomiting and hiccupping. Neuroimaging has revealed that areas with high AQP4 expression are common sites for NMO-typical lesion localization. These sites include the area postrema, where the chemo-sensitive vomiting center is located. Thus the AQP4-rich area postrema is an important first point of attack in NMOSD [11].
Unlike previous cases, serum samples of our patients were negative for anti-AQP4 antibody. Although the anti-AQP4 antibody is an important pathogen in NMOSD, some NMOSD cases cannot be confirmed using this autoantibody, even via sensitive assays. It remains unknown whether these results imply the presence of as yet unidentified novel antibodies in AQP4-Ab-negative NMO or simply a low titer or low affinity NMO-IgG/AQP4-Ab not detectable with current techniques [12]. Previous reports compared the clinical manifestations of seropositive and seronegative patients with definite NMO [13,14]. Therefore, although a positive serum test is of utmost importance for the diagnosis of NMOSD, a negative result alone cannot rule out the diagnosis. Thus, Lana-Peixoto and Callegaro [15] represented an expanded spectrum of NMO disorders, which includes patients who have optic neuritis or LETM in association with lesions typical of NMO on brain MRI, and yet are AQP4-IgG seronegative (Table 1). Brain MRI lesions typical of NMO may be considered as an alternate supportive evidence for the diagnosis of NMOSD in patients with recurrent optic neuritis, LETM, recurrent brainstem, hypothalamic, or encephalopathy symptoms and AQP4-IgG seronegative status.
In conclusion, NMOSD is an autoimmune inflammatory disease that predominantly exhibits optic neuritis and transverse myelitis. Also, brain symptoms occur frequently, not only during the course of the disease, but also preceding optic neuritis or myelitis. This case describes SIADH as an accompaniment of an NMO attack. The attack on AQP4 may affect the supra-optic and para-ventricular nucleus of the hypothalamus, causing dysfunction of ADH regulation. In addition, notwithstanding a seropositive AQP4-Ab, a seronegative result cannot rule out the diagnosis according to the expanded disease criteria. Therefore, AQP4-Ab-negative NMO can be associated with SIADH. Early recognition of the syndrome with NMO may provide important therapeutic decision-making time.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









