


Introduction: Sodium transporters in the proximal tubule
In healthy adults, the rate of renal blood flow is approximately 1.2ŌĆō1.3 L/min and that of renal plasma flow is approximately 500ŌĆō700 mL/min, corresponding to one-quarter of the cardiac output. In the glomeruli, the rate of plasma filtration into the Bowman capsule is about 100ŌĆō120 mL/min.
Almost all of the water and solutes are reabsorbed in the tubules. First, in the proximal tubule (PT), more or less 70% of the solutes (e.g., sodium, potassium, and bicarbonate) and water are reabsorbed. The remaining solutes are then reabsorbed in succeeding tubules, such as the HenleŌĆÖs loop, distal tubule, and collecting duct. Overall, almost all of the electrolytes, solutes, and water filtered in the glomeruli get reabsorbed; PT plays a significant role in this regulation of fluid homeostasis, which is, in turn, a key factor in the regulation of systemic blood pressure [1,2].
Several different types of sodium transporters are located on both the apical and basolateral sides of PT cells. At the apical surface of PT, sodium/hydrogen exchanger 3 (NHE3), sodium/glucose cotransporter 2, sodium/phosphate cotransporter type 2A, and other sodium transporters mediate sodium transport. At the basolateral surface, sodium/potassium ATPase and electrogenic sodium/bicarbonate cotransporter 1A (NBCe1A) mediate sodium transport. Among these various transporters, NBCe1A and NHE3 are the major sodium transporters in PT [2].
The NBCe1A reabsorbs sodium and bicarbonate in cooperation with the apical NHE3 and with carbonic anhydrase type 2 (CAII) in the cytosol. The importance of NBCe1A in the regulation of acid-base homeostasis is underscored by the fact that mutations of SLC4A4 (the gene encoding NBCe1) cause severe proximal renal tubular acidosis (pRTA) [3ŌĆō8]. In patients with these mutations, the pRTA condition is usually accompanied by ocular abnormalities, such as band keratopathy, glaucoma, and cataracts, and sometimes by brain calcification, mental retardation, and migraine. Functional analyses of pRTA-associated SLC4A4 mutations have revealed that reduction of NBCe1 activity by 50% or more causes severe pRTA [4,7]. Transgenic mice with targeted ablation of NBCe1 activity largely captured these human phenotypes; NBCe1-null mice produced by Shull and colleagues via targeted disruption of the Slc4a4 gene showed severe metabolic acidosis, hyponatremia, growth retardation, anemia, splenomegaly, and abnormal dentition, and they died before weaning [9]. One of NBCe1 mutations found in human, p.W516* knock-in mice also showed severe pRTA, growth retardation, ocular abnormalities, anemia, volume depletion, azotemia, dehydration, and early lethality within three weeks after birth. The administration of bicarbonate to p.W516* mice markedly prolonged their survival [10].
Up to now there have been no clear evidence that shows the direct relationship between NBCe1 function and hypertension. However, only the loss-of-function mutations of NBCe1 have been identified so far. These patients show rather hypotension, which were probably not paid attention to, because they show severe pRTA. On the other hand, the gain-of-function mutation of NBCe1 has never been found.
The significance of PT angiotensin II receptors in the regulation of systemic blood pressure
Sodium transporters in the PT are regulated by hormones, such as angiotensin II (AngII), insulin, dopamine, parathyroid hormone, nitric oxide (NO), and endothelin [2]. Among these hormones, AngII is probably the most potent stimulator.
AngII is a peptide hormone that exerts a strong pressor effect. One of its precursors is angiotensinogen, produced mainly in the liver. Angiotensinogen is subsequently converted into angiotensin I by renin in the juxtaglomerular apparatus; then, angiotensin I is further converted into AngII by angiotensin-converting enzyme.
There are two major types of AngII receptor: AT1 and AT2. In mice and rats, there are two AT1 subtypes: AT1A and AT1B. All of these receptors belong to the G protein-coupled receptor superfamily [11]. The AT1 receptors are widely expressed in all of the major organs involved in the regulation of blood pressure, such as the kidney, vasculature, heart, adrenal gland, and central nervous system [12].
Recent studies using transgenic mice by Coffman and colleagues have revealed that a subtype of AngII receptor, AT1A, in PT has a crucial role in regulating systemic blood pressure [13,14]. They investigated the importance of the different AngII receptor subtypes in the kidney, using a cross-transplantation model and showed that kidney AT1 receptors are as important as systemic AT1 receptors for the regulation of systemic blood pressure [15]. Next, using the same cross-transplantation system, they discovered that kidney AT1 receptors, not extrarenal AT1 receptors, are responsible for the pathophysiology of hypertension and cardiac hypertrophy caused by AngII [13]. Further, using mice with a specific knockout of AT1A in PT, they showed that AT1A receptors in PT regulate systemic blood pressure [14]; the knockout mice had significantly lower systemic blood pressure than the wild-type (WT) mice.
Together, the findings of Coffman and colleagues [13ŌĆō15] suggest that among the several other different organs where AT1 receptors are expressed (e.g., vasculature, heart, adrenal gland, and central nervous system), the kidney may be the most important target for the pressor effect of AngII. Moreover, AT1A in PT, in particular, seems to be responsible for the AngII effect on systemic blood pressure and for the pathogenesis of complications of hypertension, such as cardiac hypertrophy [16].
Which receptor mediates the biphasic effect of AngII?
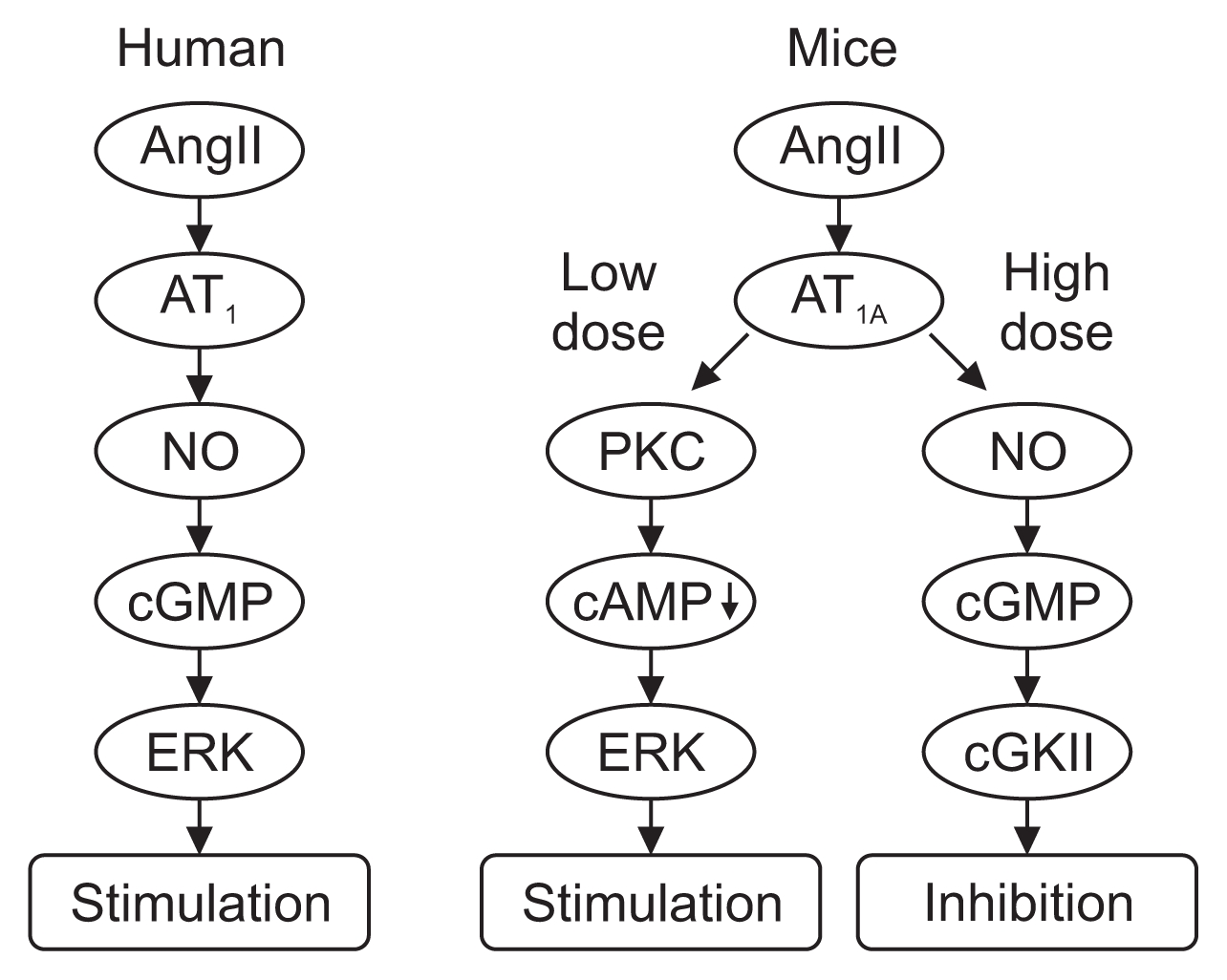
It has been known for a long time that the effect of AngII on PT Na transport is biphasic. It was shown in rats and in rabbits that AngII stimulates PT Na transport at low concentrations (10ŌłÆ11ŌĆō10ŌłÆ10 mol/L), whereas it inhibits PT Na transport at high concentrations (10ŌłÆ8ŌĆō10ŌłÆ6 mol/L) [17ŌĆō20]. However, the mechanistic details as to which receptor, AT1 or AT2, mediates this effect, had been a point of dispute. Han and colleagues [21] showed that AT1-specific antagonist losartan inhibited AngII binding to AT receptors but AT2-specific antagonist PD123,319 did not, suggesting that AT1 mediates the effect of AngII. In addition, AT1 but not AT2 was shown to be coupled with PT calcium signaling cascade [22]. However, others insisted that AT2 mediates AngII-induced reduction of bicarbonate absorption in mouse PT cells [23].
To resolve the issue of which AT receptor mediates the biphasic effect of AngII, we investigated PT basolateral transport using AT1A knockout mice. In PT of WT mice, An-gII at low concentration (10ŌłÆ10 mol/L) stimulated NBCe1 activity, whereas AngII at high concentration (10ŌłÆ6 mol/L) inhibited NBCe1 activity [24], as was previously found in rats and rabbits [17ŌĆō20]; this biphasic effect was absent in PT of AT1A knockout mice. In addition, AT1-specific antagonist valsartan applied to PT of WT mice also abolished the biphasic effect of AngII, whereas PD123,319 did not alter the biphasic effect. We also investigated apical transport and confirmed there was a biphasic effect of AngII on apical bicarbonate transport in PT of WT mice. This biphasic effect was abolished by valsartan, but not by PD123,319, and it was absent in PT of AT1A knockout mice, as had been seen on the basolateral side [25]. On the basis of these results, we concluded that the biphasic effect (stimulation at low concentration and inhibition at high concentration) of AngII on PT Na transport in mice is mediated by AT1A.
We next investigated the nature of the signal transduction cascades involved in these biphasic responses to AngII [26]. The stimulatory response was blocked by MEK/ERK inhibitor PD98059 in WT mice but not in AT1A knock-out mice. On the other hand, the inhibitory response was blocked by either AACOCF3, the cytosolic phospholipase A2 (cPLA2) inhibitor, or ketoconazole, the cytochrome P450 inhibitor; in the presence of these inhibitors, all concentrations of AngII stimulated NBCe1 activity in PT of WT mice. Additionally, in cPLA2╬▒-deficient mice, all concentrations of AngII (10ŌłÆ12ŌĆō10ŌłÆ6 mol/L) stimulated NBCe1. We also observed that arachidonic acid inhibited NBCe1 activity both in WT mice and in AT1A knockout mice [24,25]. Thus, we considered that the arachidonic acid cascade and P450 mediate the inhibitory effect of AngII on PT in mice.
Species difference in the AngII effect on the PT
Works from other groups suggested that the NO/cyclic GMP (cGMP) pathway mediates the inhibitory effect of AngII in PT of rats [27,28]; thus, we also investigated the effect of the NO/cGMP pathway on proximal transport in mice [29]. We found that NO donor sodium nitroprusside (SNP) inhibited NBCe1 activity in a dose-dependent manner; we also found that cGMP analogue 8-Br-cGMP inhibited the activity. On the other hand, NO synthase (NOS) inhibitor L-NAME, soluble guanylate cyclase (sGC) inhibitor ODQ, or cGMP-dependent kinase (cGK) inhibitor KT5823 switched the inhibitory effect of AngII to a stimulatory effect. All concentrations of AngII induced similar intensities of ERK phosphorylation in the presence of L-NAME; however, cGMP and SNP themselves did not stimulate ERK phosphorylation. These results suggest that the NO/sGC/cGMP/cGKII cascade mediates the inhibitory effect of AngII via ERK in mice.
In contrast to these findings in mice [29], markedly different results were obtained in tests of human PT. We found that AngII stimulated both NBCe1 activity and ERK phosphorylation in similar dose-dependent fashion. These effects were abolished by valsartan and PD98059, suggesting that AT1 and the MEK/ERK pathway mediate the stimulatory AngII signal. Unlike the situation in mice, both SNP and 8-Br-cGMP stimulated NBCe1 activity; in addition, both L-NAME and ODQ abolished the NBCe1-stimulatory activity of AngII, whereas cGK inhibitor KT5823 had no effect on this activity. These results indicate that in human PT the AngII signal is mediated by NO/sGC/cGMP/ERK pathway signaling and that cGKII is not involved. The reason why humans and other species have different mechanisms of PT Na transport regulation by AngII and NO has not yet been elucidated; however, the orthostatic difference, erect bipedalism in human v.s. quadrupedalism in rodents, might account for this difference [29].
In summary, in rodents and rabbits, the stimulatory effect of low-dose AngII on PT is mediated by the AT1A/PKC/cAMP/ERK pathway, while the inhibitory effect of high-dose AngII is mediated by the AT1A/NO/cGMP/cGKII pathway or by the AT1A/cPLA2/arachidonic acid pathway. In humans, AngII has a monophasic dose-dependent stimulatory effect on PT, mediated by the AT1/NO/cGMP/ERK pathway. Previous clinical data showing that NO derivatives do not lower blood pressure and can even induce sodium retention and hypertension [30,31] support our results. Fig. 1 shows the AngII effects on human and mouse PT.
Insulin and its signal transduction cascades
Insulin was first discovered as a hormone that regulates glucose homeostasis [32]. Now it is widely known that hyperinsulinemia or insulin resistance is related to metabolic syndrome, including hypertension [33]. In the latter half of this review we will at first provide the general description about the insulin signaling, followed by the effect of insulin on the PT Na reabsorption.
The insulin signaling is mediated by several signal transduction pathways. The classical PI3K-Akt pathway is probably the most important and most thoroughly investigated. In this signaling pathway, activation of PI3K via insulin receptor substrate (IRS) triggers the phosphorylation of Akt. The MEK/ERK pathway is another important insulin signaling pathway.
There are multiple IRS subtypes, among which IRS1 and IRS2 are the major two involved in insulin signaling [34]. The first IRS discovery was that of IRS1 in hepatoma cells, and IRS1 was later identified in several other tissues and organs, such as muscle, heart, liver, adipocytes, and kidney [35]. Later studies of IRS1-null mice led to the identification of an alternate IRS, designated IRS2 [36,37]. The distribution of IRS2 is similar to that of IRS1. Notwithstanding their similarities, there are some differences between IRS1 and IRS2 with respect to their tissue distributions and the way in which they contribute to glucose homeostasis. In particular, their roles in adipose tissue and hepatocytes seem to be quite different. These differences have been well investigated in knockout mice. Mice with knockout of IRS1 did not develop diabetes because IRS2 compensated for the IRS1 deficiency. Interestingly, skeletal muscle and adipose tissue of the IRS1 knockout mice showed reduced insulin-dependent glucose transport [38,39]. This may explain the reduced capacity of insulin to lower plasma glucose level in the IRS1 knockout mice. On the other hand, the IRS2 knockout mice developed peripheral insulin resistance and progressed to overt diabetes early in life, by ten weeks of age [40]. Intriguingly, in these IRS2 knockout mice, the hepatic insulin resistance induced prolonged hyperglycemia and insulin resistance in skeletal muscle [41].
Insulin resistance and tissue specificity
Insulin resistance refers to the state whereby insulin is unable to stimulate enough uptake of glucose into tissues; it is usually accompanied by hyperinsulinemia [33]. As noted above, insulin resistance can be tissue-specific, a phenomenon that seems to be partly because of differences between the roles of IRS1 and IRS2. Among the major targets of insulin, adipose tissue and liver have been the most extensively investigated with respect to mechanisms of insulin resistance and the role of IRS proteins.
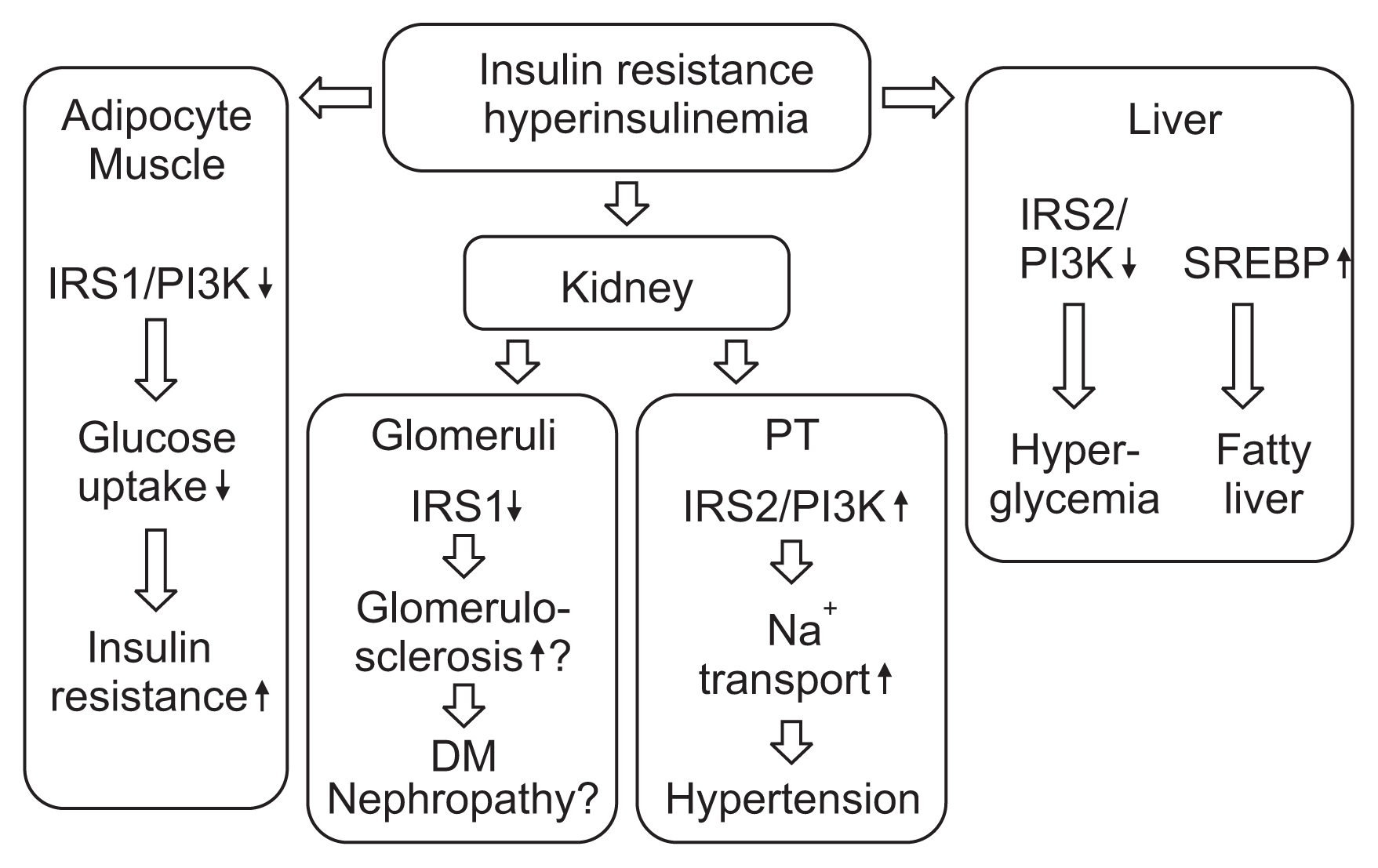
In adipose tissue, IRS1 is a major mediator of insulin signaling. In insulin resistance, the insulin signaling cascade is significantly impaired and glucose uptake into adipose tissue is attenuated. In comparison with healthy individuals, IRS1 expression is markedly reduced in the adipose tissue of insulin-resistant individuals, while IRS2 expression is unchanged [42]. As IRS2 requires a higher dose of insulin to bind to PI3K, these results suggest a mechanism for the emergence of insulin resistance in adipose tissue [42].
In the liver, on the other hand, the regulation of glucose homeostasis by IRS1 and IRS2 is different from that in adipose tissue; in the liver, IRS2 is the main contributor to the insulin signaling cascade [43,44]. Activated Akt produced through the insulin signaling cascade is thought to suppress gluconeogenesis in the liver via lowered expression of gluconeogenic enzymes (e.g., phosphoenolpyruvate carboxykinase and glucose-6-phosphatase), nuclear exclusion of forkhead box O (FoxO) protein, and inactivation of glycogen synthase kinase-3╬▓. Additionally, sterol regulatory element-binding protein 1 (SREBP-1) is known to inhibit IRS2-mediated signaling by antagonizing the effect of FoxO1 [45]. It is also known to activate the transcription of lipogenic genes [46]. This process is insulin-sensitive; hence, in hyperinsulinemia de novo fatty acid synthesis is elevated [47]. On the other hand, IRS1-mediated signaling was also shown to be essential for insulin-induced SREBP-1c expression [48]. Thus, regulation of glucose and lipid homeostasis by IRS1 and IRS2 is tissue-specific. Moreover, IRS1 and IRS2 seem to function differently in the insulin-resistant state as compared with the insulin-sensitive state.
Insulin resistance and the kidney
The kidney is also a target organ of insulin. Insulin binds to the insulin receptors (IR) throughout the nephron [49], which are essential for the proper function of the nephron, glomerulus, and tubule [50]. Previous reports have demonstrated that insulin increases sodium reabsorption [51ŌĆō53] and stimulates sodium transporters [51,52,54] in PT. This antinatriuretic effect seems to be preserved even in insulin resistance, hyperglycemia, and diabetes [55], thus perhaps contributing to hypertension, edema, and congestive heart failure in patients with diabetes.
In the glomerulus, all the three cell typesŌĆömesangial cells, glomerular endothelial cells, and podocytesŌĆöexpress IR. Among these, the podocytes are particularly important targets of insulin, as they express the highest levels of IR and IRS1 compared with mesangial cells and glomerular endothelial cells [56]. In insulin resistance, the insulin signaling cascade in the glomerulus seems to be impaired [57]. Moreover, when IR expression in podocytes is lost, symptoms of diabetic nephropathy develop [50]. Mima and colleagues [56] showed that, in a rat model of insulin resistance and diabetes, insulin-induced phosphorylation of IRS1, Akt, endothelial NOS, and glycogen synthase kinase-3╬▓ were all selectively inhibited in the glomeruli, but not in the tubules. This result suggests that IRS1 is a main contributor to the insulin signaling cascade in the glomerulus.
Chronic kidney disease (CKD) might develop as a consequence of systemic insulin resistance. Acting as the glomerular filtration barrier, podocytes are essential for maintaining glomerular function; thus, the impairment of podocyte function because of insulin signaling deficiency could be a crucial step in the progression of CKD [57].
Effect of insulin on the renal PT
It has long been known that insulin stimulates sodium transporters in PT, such as NHE3 [54], NBCe1 [58], and Na/K-ATPase [52], and enhances volume absorption [51]. Moreover, a compelling hypothesis has been proposed whereby hyperinsulinemia itself could induce hypertension by triggering renal sodium retention [59,60]. Additionally, insulin itself has been shown to have an antinatriuretic effect in humans [61].
With these facts in mind, we investigated the mechanisms underlying insulin enhancement of sodium re-absorption in PT [62]. In mouse PT, insulin at a concentration of approximately 10ŌłÆ8 mol/L stimulated NBCe1 activity. This stimulation was abolished in the presence of PI3K inhibitor, either wortmannin or LY-294002, suggesting that the stimulatory effect of insulin on NBCe1 activity in PT is mediated by PI3K. Moreover, insulin induced the phosphorylation of Akt, which was also abolished by wortmannin. Next, we used IRS1 or IRS2 knockout mice to investigate involvement of IRS1 and IRS2. In IRS1-null mice, insulin stimulated NBCe1 activity to a similar extent as in WT mice; however, in IRS2-null mice the magnitude of stimulation was approximately half that of either WT or IRS1-null mice. The magnitude of insulin-induced Akt phosphorylation in the renal cortex of IRS2-null mice was also significantly lower than that in either WT or IRS1-null mice. These results suggest that the stimulatory effect of insulin on NBCe1 in PT is mediated by the classical insulin-signaling IR/IRS2/PI3K pathway.
Effect of insulin on PT in insulin resistance and in advanced type 2 diabetes mellitus
On the basis of the foregoing evidence, we investigated the details of this insulin signaling cascade in insulin resistance [63]. We first confirmed that in Wistar rats insulin stimulates NBCe1 activity via IRS2, as it does in mice. Next, using a rat model of insulin resistance and hyperinsulinemia, the Otsuka Long-Evans Tokushima Fatty rat (OLETF) [64], we investigated whether this stimulatory effect would occur in insulin resistance. The results were intriguing; in the insulin-resistant rats, stimulation of NBCe1 in PT was preserved, while glucose uptake activity in adipose tissue was impaired, as compared with their Long-Evans Tokushima (LETO) rat counterparts. Expression of IRS1 was reduced both in PT and in fat. On the other hand, expression of IRS2 was preserved in PT but reduced in fat; similarly, phosphorylation of Akt was also preserved in PT but reduced in the fat of these insulin-resistant rats. Together, the results indicate that in PT, unlike in fat, the insulin signaling cascade via IRS2 is preserved even in insulin resistance.
We further investigated whether this stimulation of PT Na transport by insulin via IRS2 is also preserved in human insulin resistance. The results were quite similar to those observed in the OLETF rats. In the PT, as compared with individuals without insulin resistance, the stimulation of NBCe1 activity by insulin was well preserved in individuals with insulin resistance. In addition, expression of IRS1 was reduced, while expression of IRS2 was preserved in individuals with insulin resistance. The results obtained in the fat of individuals with insulin resistance were quite similar to those of OLETF rats. These results [63] suggest that in not only rat insulin resistance, but also human insulin resistance, stimulation of PT sodium transport by insulin via IRS2 is preserved, thereby enhancing sodium reabsorption in the PT.
We then investigated differences in insulin signaling between the kidney cortex and liver in Wistar rats. In the liver, feeding plus insulin significantly attenuated the expression of IRS2 from the starvation levels, whereas the expression of SREBP-1 was markedly increased from the starvation levels. In the kidney cortex, on the other hand, expression levels of SREBP-1 were unchanged between the starvation and feeding-plus-insulin conditions, respectively. In the OLETF rats, the expression of liver SREBP-1c with feeding plus insulin was much more elevated than the starvation level, whereas the expression of kidney cortex SREBP-1c was unchanged. The results suggest that, unlike in the liver, the expression of IRS2 in the kidney cortex is not affected by alteration of glucose homeostasis, such as insulin resistance.
The results thus far suggest that insulin stimulates PT sodium transport in both humans and rats, not only in insulin-sensitive individuals/animals but also in those with insulin resistance and/or hyperinsulinemia. This effect of insulin seems to be mediated by IRS2, following the classical insulin-signaling IR/IRS2/PI3K pathway. The regulation of insulin signaling in kidney PT is suggested to be different from that in liver and adipocytes.
Finally, we investigated whether the IRS2-mediated stimulation of PT Na transport by insulin was preserved in type 2 diabetes mellitus (T2DM) [65]. In the OLETF rats with T2DM and overt proteinuria, the stimulation of NBCe1 by insulin was well preserved, being similar to that of their LETO rat counterparts. Additionally, expression of both IR and IRS1 protein was impaired, while that of IRS2 protein was preserved, in diabetic OLETF rats as compared with LETO rats.
These results suggest that, in insulin resistance and T2DM, intensive glucose-lowering therapy may bring risks of sodium retention and congestive heart failure. Intensive glycemic control therapy has failed to yield apparent benefits with respect to cardiovascular events and/or mortality in large clinical trials [66ŌĆō68]. In CKD patients with T2DM, tight glycemic control, which often requires higher doses of insulin than conventional glycemic control, may increase cardiovascular risk [69]. Fig. 2 summarizes the effect of insulin, in insulin resistance, on the major insulin target tissues.
Conclusion
We have summarized here the regulation of blood pressure by renal proximal tubular transport, highlighting the major regulators of PT Na transport, namely AngII and insulin. The question of what is the primary target organ of AngII has been in dispute for a long time. A series of works by Coffman and colleagues [13ŌĆō15] revealed that the renal PT AT1A receptor is probably the primary target for AngII effects, contributing not only to fluid homeostasis but also to the regulation of blood pressure. We have further revealed that the effect of AngII on PT sodium transport is mediated by AT1 in a biphasic manner in mice: stimulation in low concentration and inhibition in high concentration. We also showed that in human PT, AngII dose-dependently and strongly stimulated the activity of NBCe1.
The particular roles of IRS1 and IRS2 depend on the tissue, and the differences become even more pronounced in insulin resistance. In adipose tissue, impairment of IRS1 signaling causes a reduction in glucose uptake by adipocytes. Hepatocytes are another important target of insulin; in insulin resistance, both IRS1 and IRS2 signaling is attenuated, resulting in hyperlipidemia and hyperglycemia.
In the kidney PT, insulin stimulates NBCe1 activity, enhancing sodium reabsorption in this segment. Our studies have revealed that IRS2 mediates the stimulation of NBCe1 activity by insulin, and that in both insulin resistance and overt diabetic nephropathy, this IRS2-mediated signaling is preserved, resulting in persistent stimulation of NBCe1 and thus enhanced sodium reabsorption.
However, the detailed mechanisms whereby insulin resistance leads to hypertension and an increase in cardiac mortality still need to be clarified. Also, the differences between the short-term and long-term modulation of blood pressure in the renal PT by AngII still need to be elucidated. Crowley and colleagues [70] have suggested that, in the long term, AngII affects the renal vascular system. We anticipate that further investigation in the future will uncover the detailed mechanisms by which AngII and insulin induce hypertension and cardiac complications through their effects on the renal PT and other tissues.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









