



Introduction
Alport syndrome (AS) is a hereditary condition characterized by progressive kidney disease, sensorineural hearing loss, and ocular abnormalities [1]. AS is caused by a defect in one of three type IV collagen alpha chains due to a pathogenic variant of the genes encoding these proteins: type IV collagen ╬▒3 chain [╬▒3(IV)], encoded by COL4A3; ╬▒4 chain [╬▒4(IV)], encoded by COL4A4; and ╬▒5 chain [╬▒5(IV)], encoded by COL4A5. There are three types of AS, depending on the mode of inheritance: X-linked AS (XLAS), autosomal recessive AS (ARAS), and autosomal dominant AS (ADAS). XLAS is caused by COL4A5 gene variants, whereas ARAS and ADAS are caused by COL4A3 or COL4A4 gene variants. Male XLAS and ARAS patients develop end-stage kidney disease (ESKD) at a mean age of around 20 to 30 years [2,-6], whereas in female XLAS and ADAS cases this occurs at around 60 to 70 years [7,-9]. Male XLAS cases show a very strong genotype-phenotype correlation, whereas this correlation is weak in ARAS cases and absent in female XLAS and ADAS cases. In addition, some XLAS cases show atypical mild phenotypes, but in most cases, the factors leading to the milder phenotypes can be clarified by identifying the genetic background, such as missense variants, in-frame deletion variants, or somatic mosaic variants [1]. One of these atypical cases yielded findings that led to the development of exon skipping therapy, a gene-targeted therapy, for male XLAS cases with truncating variants that showed severe phenotypes [10]. In this review article, we discuss the genetic background of AS, novel assays for determining the pathogenicity of variants, and current treatment options; we also introduce exon skipping therapy that we are developing and that has been confirmed to be very effective against severe XLAS cases, at least in animal models [10]. In addition, we are attempting to construct kidney organoids from induced pluripotent stem (iPS) cells, not only to identify the ╬▒5(IV) expression pattern in the glomerular basement membrane (GBM), but also in an attempt to establish a model to assess the novel treatment effects.
Molecular onset mechanisms for AS
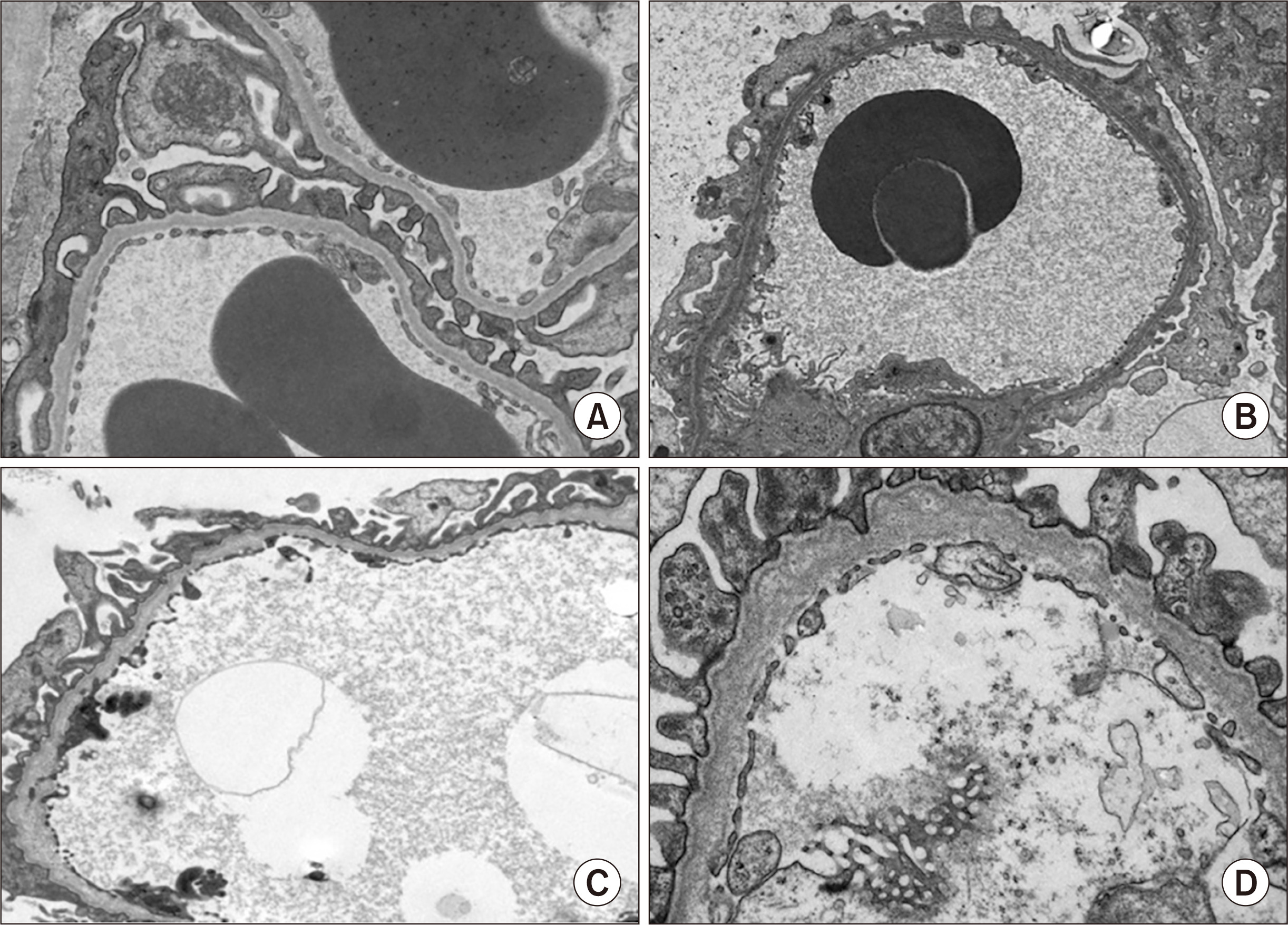
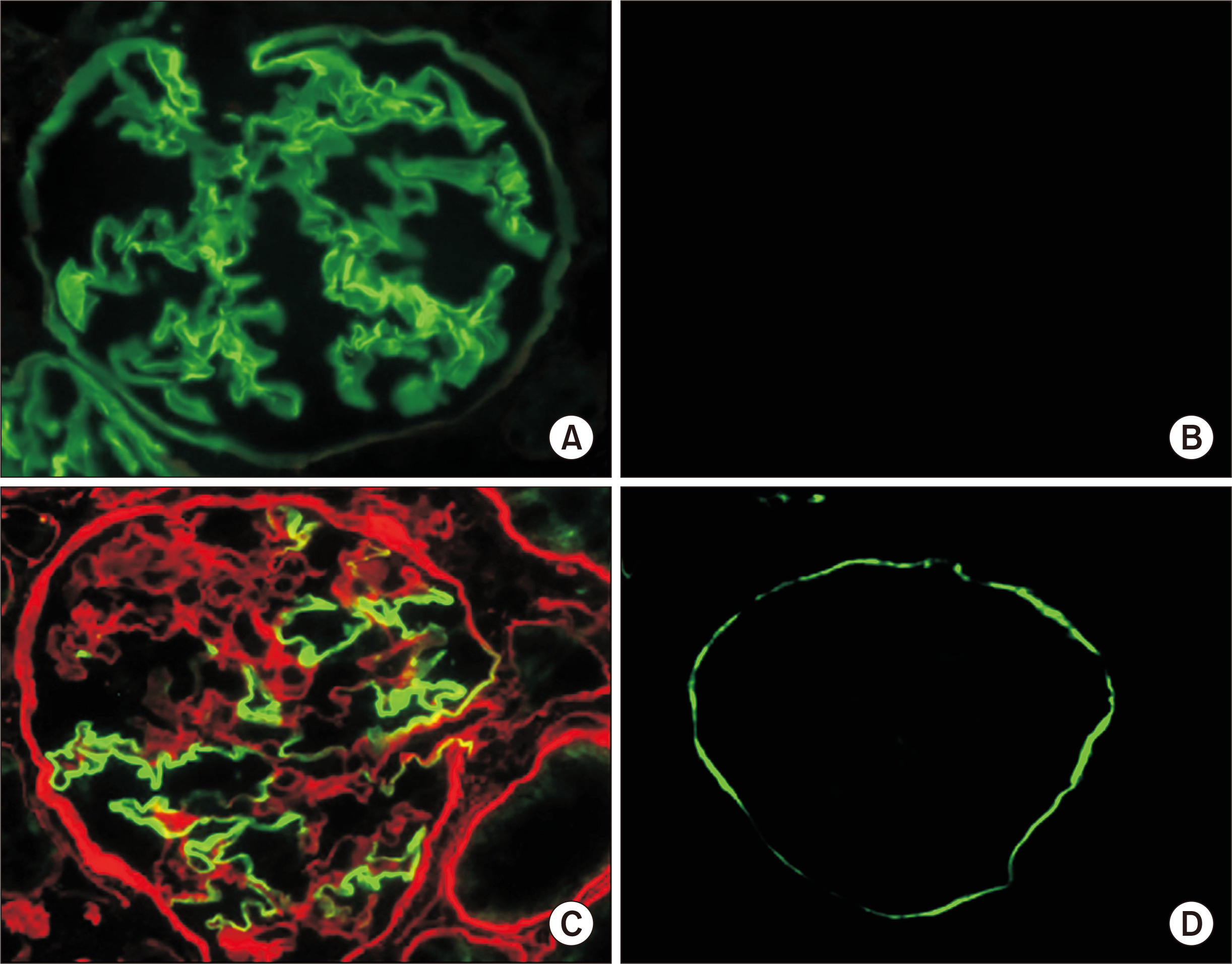
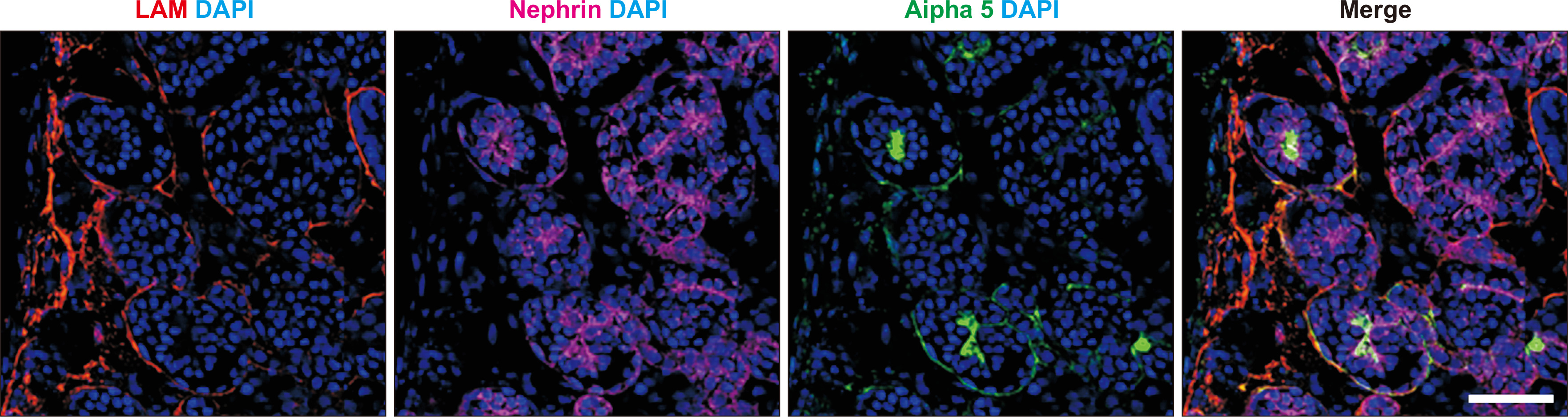
╬▒3(IV), ╬▒4(IV), and ╬▒5(IV) form a triple helix (Fig. 1A) that combines tightly with other triple helices to form the GBM. If one of the three ╬▒ chains becomes defective from a pathogenic variant of the encoding gene, the normally highly ordered GBM gradually breaks down, including splitting of the lamina densa in GBM, which is referred to as the basket weave change (Fig. 2). These changes accelerate the glomerular sclerotic changes and lead to kidney dysfunction. Immunostaining of the glomerulus revealed that ╬▒5(IV) is normally detected in both GBM and BowmanŌĆÖs capsule. However, male XLAS cases show complete negativity for ╬▒5(IV) expression, while female XLAS cases show ╬▒5(IV) expression with a mosaic pattern; ARAS cases show ╬▒5(IV) expression only in BowmanŌĆÖs capsule; and ADAS cases show normal ╬▒5(IV) expression (Fig. 3). We described the molecular mechanisms behind these expression patterns in a previous review article [1]. The ╬▒5(IV) expression patterns can be explained by the lack of production of ╬▒3(IV), ╬▒4(IV), or ╬▒5(IV), resulting in the failure of ╬▒345(IV) trimer formation (Fig. 1B). However, there are male XLAS cases with atypical expression of ╬▒5(IV). We found that ╬▒5(IV) expression was confirmed in 29% of such cases. These ╬▒5-positive cases show clearly milder phenotypes than typical cases with negative ╬▒5(IV) expression. All of these cases with ╬▒5-positive expression in GBM possessed non-truncating (missense or in-frame deletion) mutations or somatic mosaic mutations [11,12]. Therefore, for some non-truncating pathogenic variants, ╬▒345(IV) trimer can be produced, although its structure does not completely match the normal form. We discuss this point later in this review.
Overall genetic background in AS
We recently published the results of genetic testing for AS in Japan [13]. We conducted Sanger sequencing for 294 suspected AS cases and next-generation sequencing (NGS) for 147 suspected AS cases. The results showed that 239 cases in the Sanger group (81%) and 126 (86%) in the NGS group were genetically diagnosed with AS by direct sequencing methods. In addition, in 23 cases, copy number variations (CNV) were detected by pair analysis and/or multiplex ligation-dependent probe amplification (MLPA), and in eight cases, pathogenic single-base substitutions in introns were detected by RNA sequencing performed to detect aberrant splicing caused by these variants. In contrast, two cases were diagnosed with other inherited diseases: one with Brachio-oto-renal syndrome in whom nephropathy and hearing loss were observed and EYA1 gene heterozygous mutation was detected, and the other with NPHS1-related nephropathy who was showing hematuria, heavy proteinuria and pathologically thin basement membrane by electron microscopy and compound heterozygous gene variant in NPHS1. NPHS1 gene variants usually lead to a more severe phenotype of congenital nephrotic syndrome; however, some missense variants lead to milder phenotypes, such as in our case [14]. The overall diagnosis rate was 90% in this study [13].
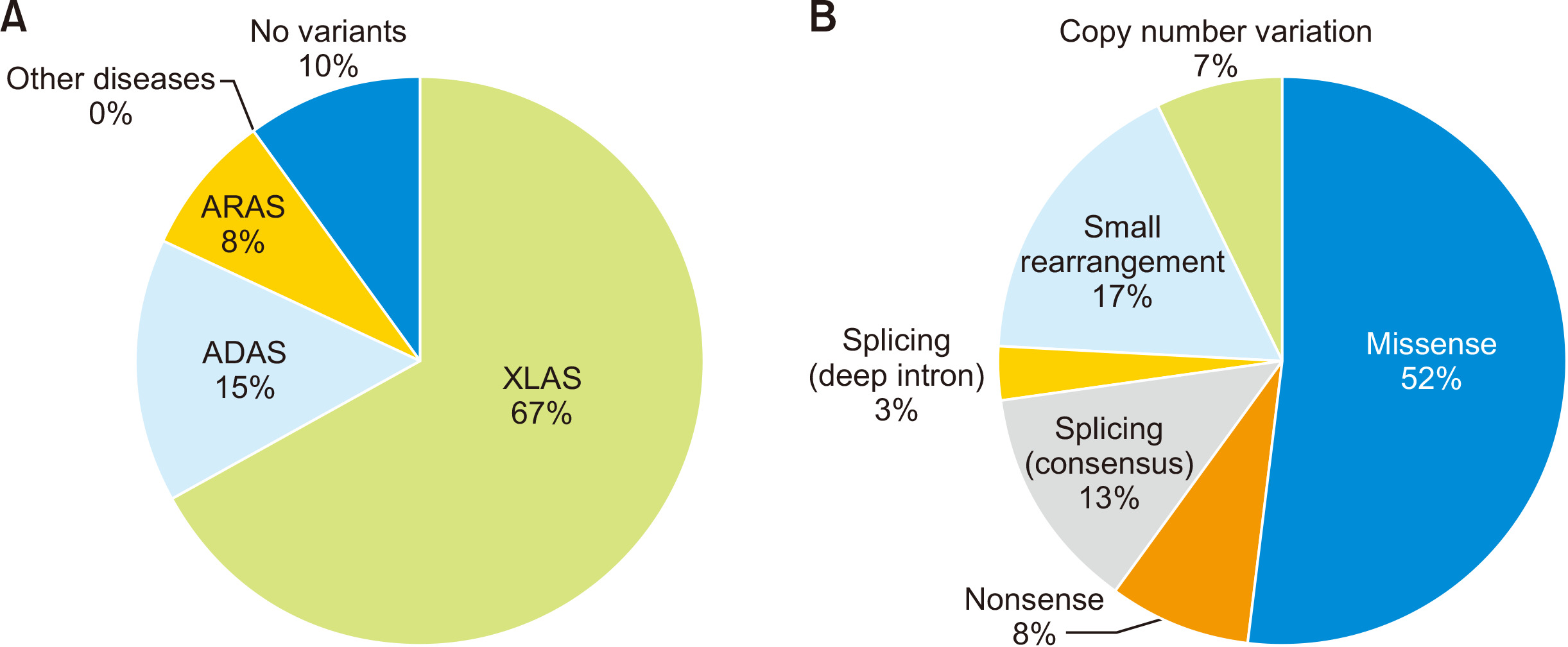
The proportions of the three modes of inheritance of XLAS, ARAS, and ADAS are 80%, 15%, and 5%, respectively [1]. However, our recent study reported proportions of 74%, 9%, and 17%, respectively (Fig. 4A included patients with no variant detection; XLAS: 67%, ADAS: 15%, ARAS: 8%, and no detection: 10%) [13]. This revealed that there are considerably more ADAS cases than previously considered. Of note, from our experience, adult cases with chronic kidney disease (CKD) accompanied by hematuria should be suspected as having ADAS when they were pathologically denied of having IgA nephropathy [15].
Among COL4A5 gene variants, 52% were missense variants, 8% were nonsense variants, 13% were splicing consensus variants, and 17% were small arrangements including insertions, deletions, and duplications. These variants can be detected by sequencing analysis. In contrast, 7% involved CNVs, usually detected by MLPA or pair analysis, and 3% involved deep intronic variants leading to aberrant splicing, as detected by RNA sequencing (Fig. 4B) [16,-19]. Pair analysis is the NGS data-dependent CNV detection method that compares per base interval read depth of the wild-type form and patient. The reduction of depth by about half in two consequent exons is suspected to be a heterozygous deletion that should be confirmed by MLPA [17].
Triple helix formation assay
In recent studies, we developed in silico and in vitro assays to examine the influence of variants in COL4A genes on the ability of the proteins to form a triple helix [10,20,21].
In silico three-dimensional (3D) structure analysis of collagen triple helix
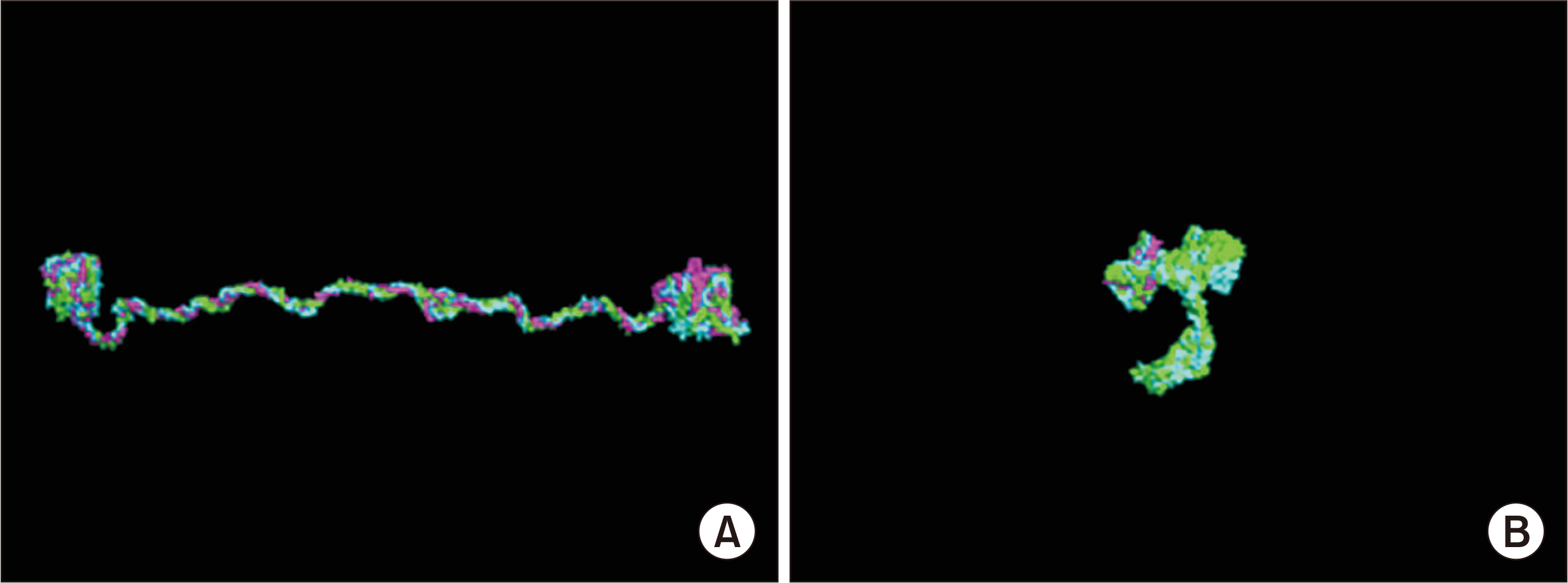
We modeled the 3D structure of the ╬▒345(IV) trimer using homology modeling [10,20,21]. Fig. 1A shows the triple helix of ╬▒345(IV) by the modeling approach. With this modeling approach, we can input information about variants and see the resulting changes of the trimer structure. Fig. 1B shows the trimer formation associated with COL4A5 nonsense mutation in exon 21, with its structure having completely collapsed. Using this approach, we showed that a noncollagenous (NC) domain encoded at the 3ŌĆÖ end of the COL4A5 gene is essential for trimer formation [10]. We are now studying whether this approach can reflect the clinical severity of pathogenic variants. For example, missense variants that change Gly to another amino acid in COL4A5 are always considered to be pathogenic. However, p.Gly953Val is considered to be nonpathogenic [22] and p.Gly1000Val leads to a very mild phenotype resembling benign familial hematuria [23]. We are now attempting to reveal these differences using this approach.
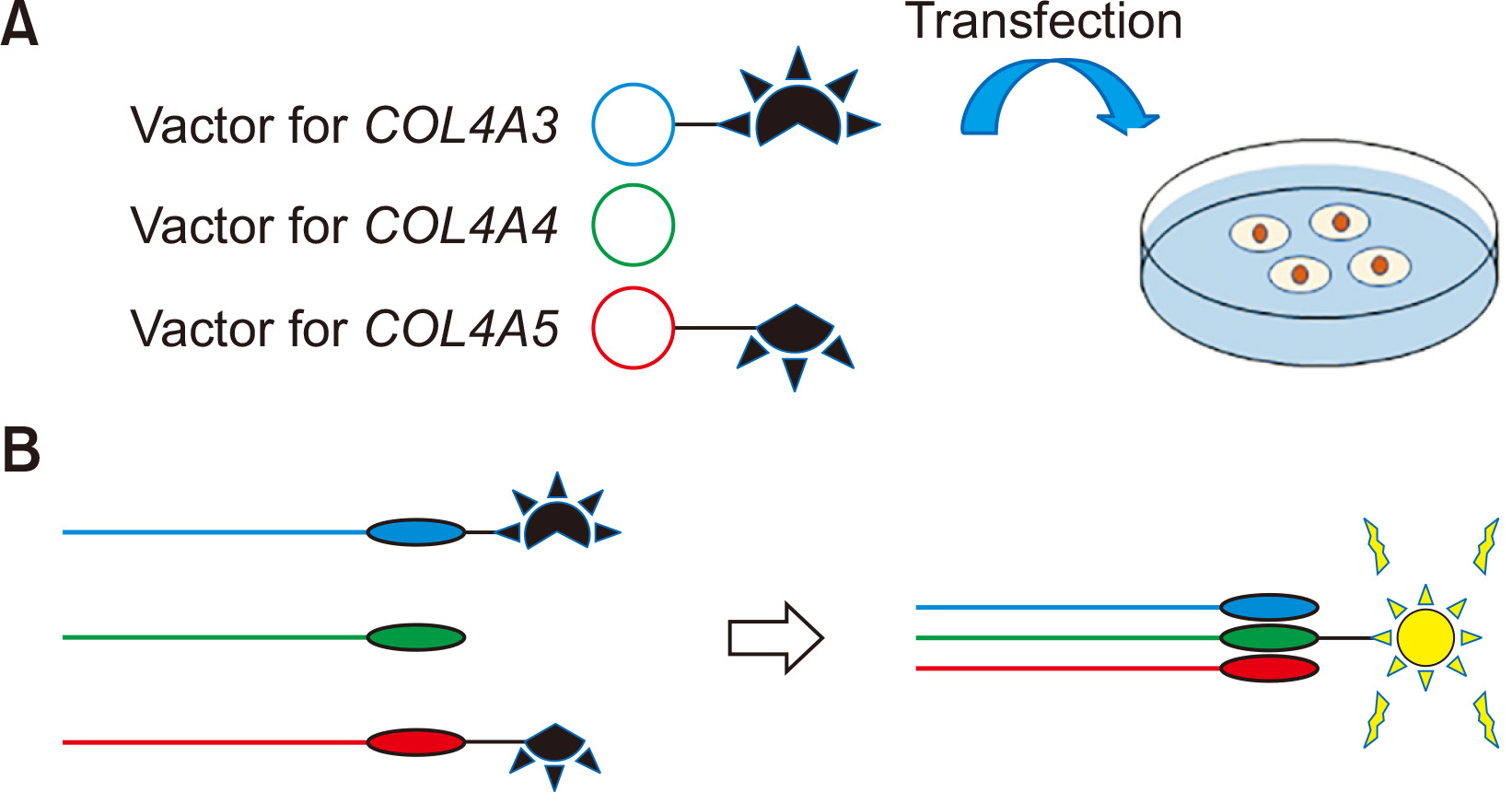
In vitro split luciferase-based trimer formation of ╬▒345(IV) protein assay
We have also established a split nanoluciferase (NanoLuc) complementation system to examine the formation of the ╬▒345(IV) trimer in vitro [20,21]. This system is composed of a large fragment and a small fragment of NanoLuc luciferase. Luminescence is observed when the split NanoLuc-tagged proteins interact with large and small fragments. We constructed plasmids containing full length of cDNA for COL4A3, COL4A4, and COL4A5, separately, and C-terminal was tagged by a small fragment for COL4A3 and by a large fragment for COL4A5 (Fig. 5A). These plasmids are transfected into HEK293T cells and luminescence is detected with high sensitivity from the trimer of ╬▒345(IV), but not from the homodimer- or heterodimer-expressing cells or supernatant (Fig. 5B). Using this assay, we revealed a strong correlation of the cell-based a345(IV) trimer formation with clinical severity [20] and anticipate the ability to distinguish pathogenic variants from nonpathogenic ones or predict the kidney prognosis. We also used this assay to show that ╬▒5(IV) with missense variants can form the ╬▒345(IV) trimer and be excreted from cells. These results are compatible with the finding that ╬▒5(IV) is expressed in some male XLAS cases with missense variants that tend to show milder phenotypes [11,12].
Generation of kidney organoids from patientsŌĆÖ iPS cells
As is caused by genetic mutations of collagen genes, it is possible to reproduce the phenotypes of AS in kidney generated from AS patient-derived iPS cells [10]. Currently, there are protocols in which human iPS cells are differentiated into self-organizing kidney-like tissue in vitro, so-called kidney organoids [24,-26]. In our protocol, kidney organoids include nephrons, collecting ducts, renal stromal cells, and endothelium [27] and develop GBM and slit-diaphragm structures in glomeruli when they are cultured in vivo by transplantation into animals [28]. We also confirmed the expression of ╬▒5(IV) in GBM of kidney organoids (Fig. 6). CRISPR/Cas9 genome-editing technology enables scientists to rapidly and reliably create desired genetic mutations of collagen genes in human iPS cells or to correct causative gene mutations in AS patient-derived iPS cells to obtain isogenic control cell lines. However, to the best of our knowledge, there have been no reported studies on AS in which degraded GBM was successfully reproduced in kidney organoids using patientsŌĆÖ iPS cells. However, such de novo technologies will enable us to investigate the development of AS in vitro.
In vitro splicing assay for detecting splicing abnormalities
RNA sequencing is often difficult because of the low expression levels of transcripts in peripheral leukocytes, fragility of transcripts, or reduced transcript expression because of nonsense-mediated decay for truncating variants. Recently, we and other groups developed an in vitro splicing assay using a minigene construct to detect aberrant splicing caused by variants in COL4A5 (Fig. 7) [16,29,-31]. This assay can easily detect the pathogenicity of single-base substitutions causing aberrant splicing in exons and introns, such as synonymous variants or variants outside of the intronic consensus sequences [16,30,32,33]. Here we describe one example of such detection (Fig. 7) in a XLAS case with a single-base substitution of IVS X+5G > A (the fifth nucleotide in intron X is mutated from G to A). Without transcript analysis, we cannot determine whether this variant is pathogenic. Thus, we can construct a vector containing a promoter, Exon A, and Exon B; in addition, a sequence containing exon X in conjunction with nearby intronic sequences harboring wild-type or the variant is inserted between Exons A and B. This vector is called a minigene construct. This vector is transfected into cultured cells and subsequently produces transcripts. The transcripts are extracted and reverse transcript polymerase change reaction is conducted to check whether the variant leads to aberrant splicing. This assay is very effective for screening the pathogenicity of splicing variants [16].
GenotypeŌĆōphenotype correlations in AS
Male XLAS
Three reports have described the correlations between genotype and phenotype in male XLAS patients [2,-4]. All of these reports describe common results of cases with truncating variants (nonsense, small rearrangement, and large rearrangement) that developed ESKD more than 10 years earlier than cases with non-truncating variants (missense and in-frame small deletion). In addition, with small deletion variants in which the number of deleted nucleotides is a multiple of 3, the cases tend to show milder phenotypes because each triplet of nucleotides encodes an amino acid. When the number of deleted nucleotides is a multiple of 3, an in-frame deletion occurs and the rest of the amino acid sequence remains unchanged. In contrast, cases with splice site variants have been reported to show intermediate severity of developing ESKD at the median age between that for truncating and non-truncating variants [2,-4]. We further investigated the correlation of phenotypes and the number of deleted nucleotides at the transcript level, concentrating on splicing variants. For all cases suspected of having aberrant splicing due to splicing variants, we conducted RNA sequencing and revealed the aberrant splicing. We divided the cases into two groups of truncating mutations (in which the deleted nucleotide number was not a multiple of 3; n = 21, from 14 families) and non-truncating mutations (in which the deleted nucleotide number was multiple of 3; n = 25, from 15 families) at the transcript level. The results showed that the median age for developing ESRD was 20 years for patients with truncating mutations and 29 years for those with non-truncating ones (P = 0.001) [16]. These results suggest that, even in cases with splicing abnormalities, there is a strong genotype-phenotype correlation.
Female XLAS
Regarding female XLAS cases, no genotype-phenotype correlations were observed in two previous studies [7,9]. Some findings suggested that an uneven pattern of X-chromosome inactivation (i.e., skewed X-chromosome inactivation) would determine the severity of XLAS in females [34,35]. However, to date, no study has systematically demonstrated this correlation.
ARAS
There are two previous reports and one systemic review article discussing the genotype-phenotype correlation in ARAS [5,6,36]. Storey et al [36] reported that patients with truncating mutations in at least one allele showed a more severe phenotype with early onset of renal failure compared with patients without truncating mutations. However, our group reported that no genotype-phenotype correlation was observed in a Japanese ARAS cohort [6]. Finally, Lee et al [5] conducted a systematic review of 148 previously reported cases and concluded that there was a genotype-phenotype correlation according to the number of missense mutations. Patients with two missense mutations had delayed onset of ESKD and rarely showed sensorineural hearing loss.
ADAS
To the best of our knowledge, no genotype-phenotype correlations have yet been observed in ADAS. Even within one family sharing the same variant, the clinical severity differed significantly [8]. In a recent study with a large cohort of CKD patients, COL4A3 or COL4A4 gene variants contributed to 16% of 312 cases with CKD [37]. Although we reported that 17% of the AS cases are ADAS, the cohort study suggested that far more CKD cases caused by ADAS are undiagnosed. The diagnosis of ADAS is difficult as such cases always lack pathological features typical of AS [15]. Therefore, it is important for nephrologists to recognize this condition and perform genetic analysis for appropriate patients. The factors determining the severity of ADAS are still unclear and can include modifier genes, other coinciding renal diseases, or other acquired factors such as hypertension, diabetes, smoking, and obesity.
The disease spectrum of ADAS is still currently under discussion. Historically, cases with only hematuria and having heterozygous variants in either COL4A3 or COL4A4 were classified with thin basement membrane disease (TBMD). In fact, most of these cases will not develop ESKD and the clinical course is benign, which is different from conventional AS. However, with the development of recent molecular diagnostic techniques, many CKD cases were revealed to be heterozygous COL4A3 or COL4A4 gene variant carriers [15,37] and they were not always benign. In addition, some of the cases diagnosed with TBMD in their early life will develop into ESKD in their later life. Therefore, recent review articles have suggested that all cases with variant carriers in these genes should be classified into autosomal AS [1,38,39]. This classification can avoid ADAS patients missing initiation of treatment.
Treatment
There is currently no radical therapy for AS; however, treatment by renin-angiotensin system inhibitors (RAS inhibitors) has been performed to reduce proteinuria and delay progression to renal failure using nephroprotective drugs. The results of two randomized controlled trials (RCTs) revealed the reduction of urine protein levels by angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers in AS [40,41]. A large retrospective study reported that ACEIs can delay the progression to ESRD in AS [42]. The results of a RCT (EARLY PRO-TECT ALPORT study) confirming the nephroprotective effects have recently been published. These results showed that ramipril treatment reduced the risk of disease progression by almost half (hazard ratio, 0.51) [43]. The study strongly recommended treatment with an RAS inhibitor for AS.
Some new drugs for AS have entered clinical trials. Bardoxolone methyl is in a phase II/III trial (CARDINAL study). Bardoxolone methyl works as an activator for the KEAP1-Nrf2 pathway and blocks the NF-╬║B pathway, activating many anti-inflammatory or antioxidant genes. A recent RCT for diabetic kidney disease patients as a phase 2 clinical trial (TSUBAKI study) showed a significant increase of measured glomerular filtration rate [44]. RG-012 (also known as lademirsen, which interferes with microRNA-21 (miR-21) interference) is under a phase II trial. miR-21 is involved in the progression of fibrogenic diseases. Inhibiting miR-21 by oligonucleotides improved the survival and histological findings in AS mice [45].
Exon skipping therapy
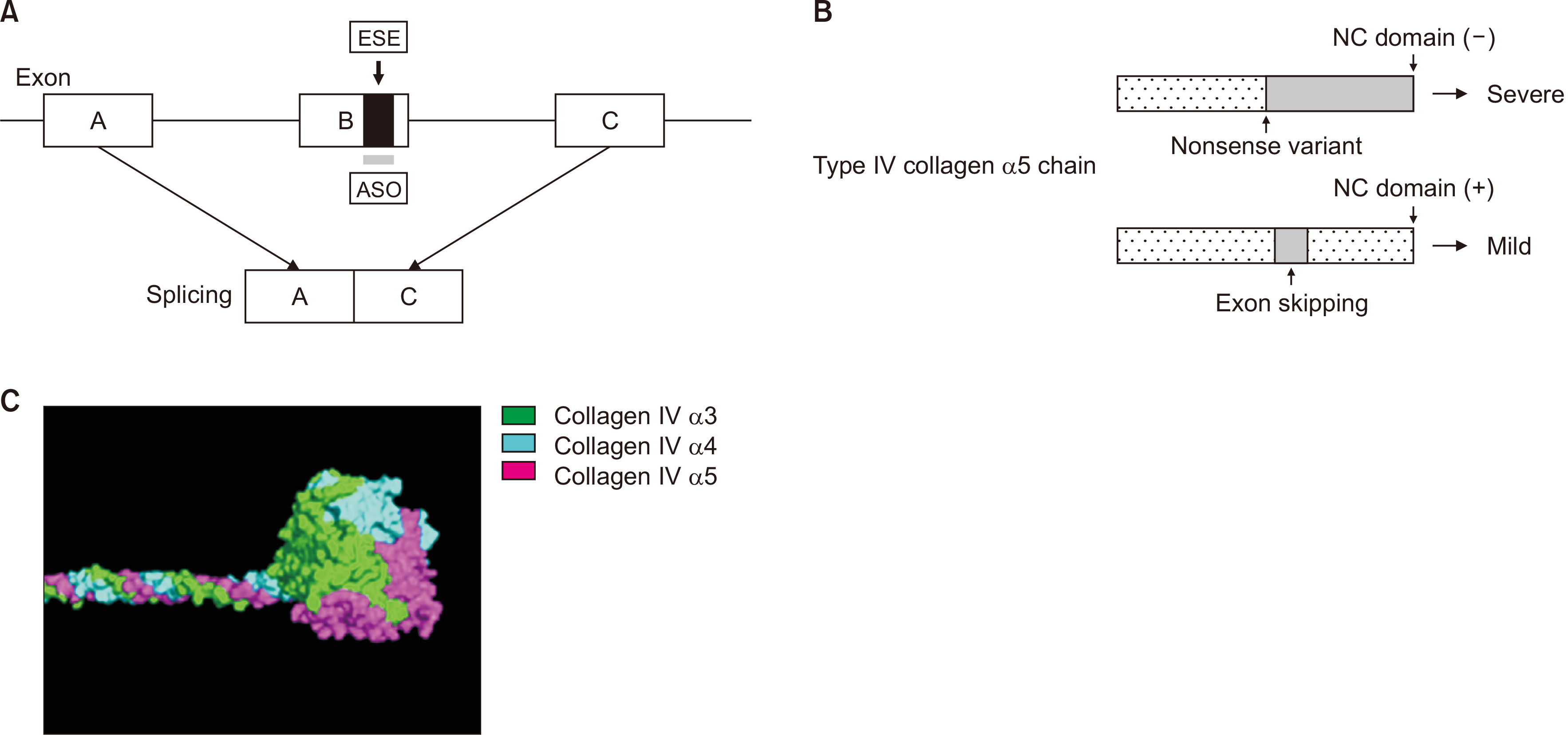
Our group is developing exon skipping therapy using antisense oligonucleotide (ASO) for treatment of severe male XLAS cases. This approach replaces the truncating variants with a non-truncating in-frame deletion mutation at the transcript level, which leads to milder phenotypes in AS. ASO binds to the exonic splicing enhancer region and, as a result, this exon is not recognized as an exon in the splicing process, leading to exon skipping (Fig. 8A). As described above, when the number of skipped nucleotides is a multiple of 3, the severe phenotype from a nonsense mutation is changed to a milder phenotype from an in-frame deletion. Among the exons in the collagenous domain of the COL4A5 gene (exons 3 to 46), 35 exons out of 44 consist of nucleotides that are a multiple of 3. Therefore, theoretically, if patients have truncating variants in one of these exons, exon skipping therapy could replace truncating variants with non-truncating variants. In these cases, exon skipping therapy would lead to establishment of the NC domain in ╬▒5(IV) (Fig. 8B), which allows the formation of a trimer with ╬▒3(IV) and ╬▒4(IV), leading to a milder phenotype (Fig. 8C). We conducted an in vivo study using an AS mouse model possessing a nonsense mutation in exon 21 of col4a5 [46] and treated mice with ASO leading to exon 21 (84 bp) skipping. This treatment had a remarkable effect of improving the pathological findings with the recovery of ╬▒5(IV) chain expression, reduced urine protein excretion level, delayed development of ESKD, and extension of survival [10]. To date, we have only developed ASO for exon 21, so further work is needed to develop this therapy to target other exons. We are currently attempting to establish a model to assess the effect of this treatment for other exons using kidney organoids. We confirmed that organoids from a patientŌĆÖs iPS cells with a truncating variant in COL4A5 gene do not express ╬▒5(IV). Our experiments to determine when and how often ASO should be injected to AS patients are ongoing. We will then conduct safety evaluation tests using animals and proceed to a clinical trial.
Conclusion
Recent developments in genetic studies and related in silico and in vitro analyses in AS have led to accurate diagnoses, investigation of the genetic background including genotype-phenotype correlation, and the development of novel therapies. Additional work in this field could help AS patients by further improving their quality of life.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









