


Introduction
The prevalences of obesity and diabetes continue to increase dramatically in many countries. Obesity, especially visceral obesity, causes insulin resistance and is associated with dyslipidemia, impaired glucose metabolism, and hypertension, all of which exacerbate atherosclerosis [1]. One recently proposed, plausible mechanism is that factors known as adipocytokines, which are produced by adipose tissue in obesity, can directly impact the atherogenic environment of the vessel wall by regulating gene expression and function in endothelial, arterial smooth muscle, and macrophage cells. These adipocytokines include tumor necrosis factor alpha (TNF-α), leptin, adiponectin, resistin, monocyte chemotactic peptide-1 (MCP-1), and plasminogen activator inhibitor-1 (PAI-1), as well as free fatty acids [2]. Reduction of visceral fat mass leads to amelioration of these risk factors and potentially prevents cardiovascular events.
Atherosclerosis is a chronic inflammatory condition of the arterial wall and causes cardiovascular complications such as ischemic heart disease, stroke, and peripheral arterial disease. Vascular smooth muscle cells (VSMCs) within the media of arteries are important in the pathogenesis of atherosclerosis. These cells respond to various cytokines and growth factors to migrate, proliferate, and produce extracellular matrix [3].
The ob gene product, leptin, is secreted mainly by adipose tissue and acts through its receptor, OB-R [4]. In previous studies, leptin has been shown to induce the proliferation of VSMC [5], [6], and this process is important in the development of atherosclerosis. Hyperleptinemia, which often coexists with diabetes and metabolic syndrome, is an independent risk factor for the progression of coronary artery disease. However, there is conflicting evidence about the exact role of leptin in the pathogenesis of atherosclerosis.
Obesity induces phenotypic changes in adipocytes, such as hypertrophy, and induces an inflammatory response in adipocytes in an autocrine or paracrine fashion to impair adipocyte function, including insulin signaling [7]. Several studies have reported that overexpression of MCP-1 induces macrophage recruitment in adipose tissue and insulin resistance [8], [9]. VSMCs express chemokine (C-C motif) receptor 2 (CCR2), the primary receptor for MCP-1, causing the expression of inflammatory genes and impaired uptake of insulin-dependent glucose [10]. MCP-1 (also known as chemokine CCL2) is one of the key cytokines that contribute to atherosclerosis by inducing monocyte trafficking and remodeling of the extracellular matrix [11], [12], [13]. Taken together, these results indicate the possibility of an expanded role of MCP-1 in vascular inflammation in metabolic syndrome. Therefore, the MCP-1/CCR2 pathway might play an important role in the pathogenesis of atherosclerosis by macrophage infiltration into VSMCs, leading to vascular inflammatory consequences.
This study was performed to investigate whether alterations of leptin levels in plasma are related to the state of systemic inflammation and severity of nephropathy in type 2 diabetic participants. In addition, we want to investigate whether MCP-1 synthesis was increased under high leptin conditions and leptin stimulation activated mitogen-activated protein kinase(MAPK) pathway in cultured VSMCs.
Methods
Study participants
A total of 126 type 2 diabetic participants and 37 healthy controls were enrolled in the study. All participants had been treated at an outpatient clinic and were recruited over 12 months. The healthy controls were chosen from an epidemiologic study conducted at the Ansan Cohort Center, which was held in 1998 in Ansan-si, Gyeonggi-do, South Korea. For the control group, age, and sex-matched participants who had no known disease history and no abnormal laboratory findings were chosen. The diagnosis of type 2 diabetes mellitus was made according to the Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus [14]. Participants had to fulfill three additional criteria for inclusion: no episode of ketoacidosis, no ketonuria, and insulin therapy initiated after at least 5 years of known disease. Participants with non-diabetic kidney diseases, and participants with azotemia (serum creatinine >1.6 mg/dL) were excluded. Participants were instructed not to make lifestyle changes or changes to their oral hypoglycemic or/and insulin therapy during the study period. Ethical approval was obtained from the Korea University Institutional Review Board, and written informed consent was obtained from all study participants.
Among the diabetic participants, 74 had hypertension. All hypertensive diabetic participants were being treated with antihypertensive medication, 61 participants were taking an angiotensin-converting enzyme inhibitor or an angiotensin II receptor antagonist with or without other antihypertensive medications. The other 13 participants who were not receiving a reninangiotensin system (RAS) blockade were being treated with calcium channel blockers, or alpha or beta blockers. Venous blood was taken from all participants after overnight fasting. Fasting blood glucose and postprandial 2-hour glucose levels were measured using the hexokinase method. Total protein, albumin, hemoglobin, creatinine, total cholesterol, triglyceride, hsCRP, and high-density lipoprotein (HDL) cholesterol levels were also measured. Hemoglobin A1c (HbA1c) was measured by high performance liquid chromatography (HPLC). The homeostasis model assessment of insulin resistance (HOMA-IR) was calculated using the standard formula. A 24-hour urine collection was performed for the determination of urinary albumin excretion and creatinine clearance. Urinary albumin excretion was measured by radioimmunoassay (Immunotech, France) at a sensitivity of 0.5 mg/L. Plasma and urinary creatinine levels were measured using the modified Jaffe method. Creatinine clearance was calculated using the morning creatinine level and was standardized for a body surface area using the standard formula. Plasma MCP-1 and leptin levels and secreted MCP-1 from culture supernatants were measured using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA). The diabetic participants were divided into three groups: a normal albuminuric diabetic group with a 24-hour urinary albumin excretion (UAE) lower than 30 mg in two or more urine samples and no more than one value greater than or equal to 30 mg (n=40); a microalbuminuric diabetic group with a 24-hour UAE in the range of 30–299 mg in at least two urine samples (n=41); and an overt proteinuria group of proteinuric diabetic participants, defined as having a 24-hour UAE greater than or equal to 300 mg (n=35).
Culture and experimental conditions for VSMCs
A rat vascular smooth muscle cell line (CRL-2018) was obtained from ATCC (Manassas, VA, USA). Cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 100 u/mL antibiotics at 37 °C under a humidified 5% CO2 atmosphere. Cultures were fed every 2–3 days during growth and every 2 days after confluence. Cells were used at 10 passages in this study. VSMCs were cultivated on 100 mm dishes and serum-restricted for 24 hours to determine whether leptin could directly increase MCP-1 synthesis. Afterward, different concentrations of recombinant rat leptin (Sigma-Aldrich, St. Louis, MO, USA) were added to culture media at final concentrations of 1 ng/mL, 10 ng/mL, and 100 ng/mL. Three or 12 hours later, the media were collected, and the cells were scraped from the dishes in the presence of extraction buffer (20 mM Tris–HCl, pH 7.4, 10 mM ethylenediaminetetraacetic acid, 5 mM ethylene glycol tetraacetate, 5 mM β-mercaptoethanol, 50 μg/mL phenylmethyl sulfonyl fluoride, 10 mM benzamidine, and 0.1 μg/mL aprotinin) and homogenized. Cell lysates and conditioned media were centrifuged at 15,000 rpm for 10 min at 4 °C, and the supernatants were collected. To define the mechanism of leptin-mediated MCP-1 synthesis and whether or not it is mediated by the MAPK/extracellular signal–regulated kinase (ERK) pathway, a selective MAPK kinase (MEK) inhibitor, PD98059 (Sigma-Aldrich, St. Louis, MO, USA), was added to the cells at a concentration of 10 μM 1 hour before treatment with leptin. Next, we performed another experiment to determine the effects of high glucose and leptin stimulation on the MAPK signaling pathway. Subconfluent VSMCs were cultured for 24 hours in a medium containing 5 mmol/L or 30 mmol/L of D-glucose. The cells were then treated with leptin at final concentrations of 1 ng/mL, 10 ng/mL, and 100 ng/mL, and harvested after 6 hours. In addition, we investigated whether leptin stimulation induces the sequential activation of the MAPK pathway, as occurs with the MEK, MAPK (ERK), and E26 (ETS)-like transcription factor (ELK) pathways. Since leptin increased maximal MCP-1 production at a final concentration of 100 ng/mL, leptin was added to the VSMCs at final concentrations of 100 ng/mL, and cells were harvested at 30 minutes, 1 hour, 3 hours, 6 hours, and 12 hours, and then the proteins were extracted. To avoid any confounding effects of serum on the MAPK pathway, all experiments were performed in serum-free media. All experimental groups were cultured in triplicate. The results were representative of those from three independent experiments.
Reverse transcription/polymerase chain reaction
VSMCs were resuspended in Trizol reagent (Invitrogen, Carlsbad, CA, USA) containing RNase inhibitor. Reverse transcription (RT) was performed using an RT polymerase chain reaction (PCR) kit (PerkinElmer, Foster City, CA, USA). A total of 1 μg of RNA and oligo-(dT)12 primers were used. The reaction mixture was incubated for 60 min at 42 °C, and was then heated for 7 min at 90 °C in a thermocycler (Crocodile III, Oncor Co., Japan). After cDNA synthesis by RT, PCR amplification was done. For MCP-1, the sense primer was 5′ GAC CTG TTT GTC CGT AAG GC 3′, and the antisense primer was 5′ GAC CTG TTT GCA ACG GGC TG 3′. The mixture was heated at 94 °C for 30 seconds, at 55 °C for 30 s, at 72 °C for 30 seconds, and this was repeated by 38 cycles. For ERK1/2, the sense primer was 3′ CTT CCT CTA CCA GAT CCT CC 5′, and the antisense primer was 3′ GTC AAG AGC TTT GGA GTC AG 5′. The mixture was heated at 94 °C for 1 min, at 57 °C for 1 min, at 72 °C for 1 min, and this was repeated by 36 cycles. The PCR data were expressed in relative values as MCP-1/β-actin, Erk/β-actin.
Western blot analysis
Cells were lysed in lysis buffer. The protein was electrophoresed on a 10% SDS–PAGE minigel under denaturing conditions. The protein was transferred onto a nitrocellulose membrane. Then, separate reactions were conducted with rabbit anti-MEK antibody, anti-p-MEK antibody, anti-ERK1/2 antibody, anti-p-ERK1/2 antibody, anti-ELK antibody, and anti-p-ELK antibody (New England Biolabs, Inc., Beverly, MA, USA) diluted 1:1000 applied to the membrane. The filter was then washed four times with PBST and incubated with horseradish peroxidase-conjugated secondary antibody diluted 1:1000 for 60 min at room temperature. The detection of specific signals was performed using the ECL method. Equal amounts of protein loading were confirmed by Coomassie blue staining of the gel.
Statistical analysis
For parametrically distributed data, we used Student's unpaired t-tests and analysis of variance (ANOVA) for comparisons of quantitative variables, and chi-square tests for comparisons of proportions. Plasma leptin, MCP-1, and UAE levels were positively skewed with a high frequency of low values. Because data transformation would not have normalized this distribution, we used non-parametric statistical methods. For the analysis of in vitro data, a nonparametric analysis was used due to the small sample. A Kruskall-Wallis test was used for comparison of more than two groups, followed by a Mann-Whitney U test, using a microcomputer-assisted program with SPSS for Windows 10.0 (SPSS Inc., Chicago, IL, USA). Correlations between plasma leptin levels and biochemical parameters were examined using Spearman's rank correlation and by stepwise multiple regression analysis. P values less than 0.05 were considered statistically significant. Results were expressed as mean±SEM.
Results
Clinical characteristics of the study sample
Table 1 shows the baseline clinical characteristics of the study sample. There were no significant differences between the participants and controls for age, sex, body weight, hemoglobin, albumin, and creatinine levels. These parameters were similar among the diabetic participants based on nephropathy status. There was no significant difference in the proportion of patients with previous history of cardiovascular disease or cerebrovascular diseases among diabetic patients (Table 1). The control group had a lower mean body mass index (BMI), fasting plasma glucose, postprandial 2-hour plasma glucose, total cholesterol, triglyceride, and hsCRP levels, systolic and diastolic blood pressure, UAE rate, and higher creatinine clearance than the diabetic groups. Among the diabetic participants, there were no significant differences in body weight, BMI, or systolic and diastolic blood pressure. The overt proteinuria group had higher UAE values and lower creatinine clearance than the normal and microalbuminuria groups. The normal albuminuric group showed higher levels of fasting plasma glucose and postprandial 2-hour plasma glucose levels than the microalbuminuria and overt proteinuria groups.
Plasma concentrations of leptin and MCP-1
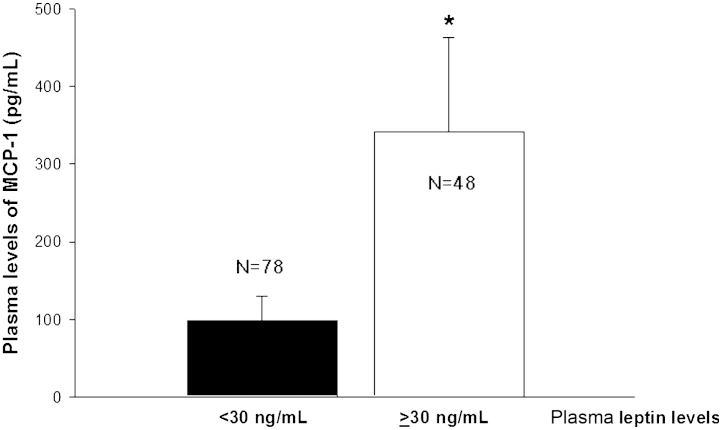
Plasma leptin levels were markedly elevated in the type 2 diabetic participants compared to the controls even in the normal albuminuric group (Table 1). Interestingly, plasma leptin levels were significantly higher in the overt proteinuria group than in the normal and microalbuminuria groups. Plasma MCP-1 concentrations were also significantly higher in the diabetic group than the control group. In the diabetic group, plasma MCP-1 levels increased according to the state of diabetic nephropathy. Since median value of plasma leptin level was 30 ng/mL, we reanalyzed the plasma MCP-1 levels based on the plasma leptin concentration using a cutoff value of 30 ng/mL. When we compared MCP-1 levels after adjusting for the four groups according to plasma leptin levels, participants who had leptin levels over 30 ng/mL had showed markedly higher levels of MCP-1 than participants with leptin levels below 30 ng/mL (Fig. 1). Table 2 shows the correlation analysis results between plasma leptin levels and various clinical parameters in the study participants. Plasma leptin levels were positively correlated with body mass index, fasting and postprandial blood glucose, HbA1c, total cholesterol, UAE, hsCRP, and plasma MCP-1 levels, and negatively correlated with creatinine clearance. However, plasma leptin concentrations did not show a significant relationship with other risk factors, including body weight, triglyceride levels, creatinine values, and systolic and diastolic blood pressure. By stepwise multiple regression analysis, plasma MCP-1 levels as a dependent variable at a cutoff value of 100 pg/mL, plasma leptin levels was only included in the model (betas±SE=−0.048±0.001, P<0.001).
MCP-1 mRNA and protein expression in cultured VSMCs in response to leptin
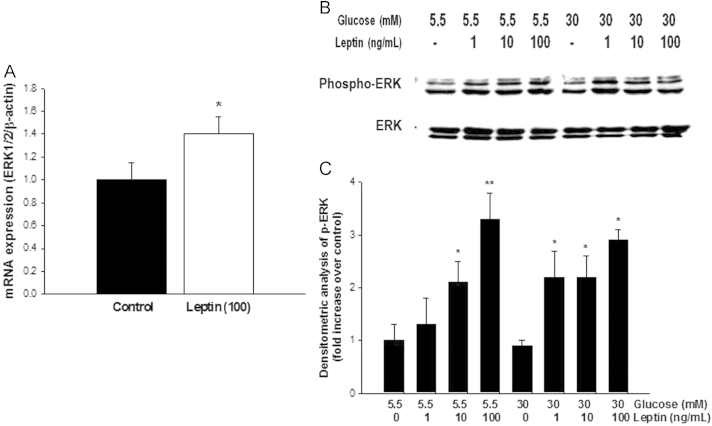
Since plasma leptin levels appear to be the most significant independent factor for MCP-1 plasma levels, we next observed the effect of leptin stimulation on MCP-1 production. As shown in Fig. 2, MCP-1 mRNA expression significantly increased after leptin stimulation in a concentration-dependent manner, except at 1 ng/mL concentration. Interestingly, selective MEK inhibitor PD98059, significantly inhibited leptin-induced MCP-1 gene expression. Based on gene expression pattern, secreted MCP-1 protein also showed a similar tendency (Fig. 2). MCP-1 secretion also time-dependently increased from 3–12 hours, and prior treatment with PD98059 also showed a significant decrease in MCP-1 production (Fig. 2).
Effects of leptin on the activation of MAPK
Since MEK inhibitor PD98059 significantly suppressed leptin-induced MCP-1 production, we further evaluated whether MCP-1 production induced by leptin depends on the MAPK pathway. We first examined the activation of MAPK (ERK) in response to leptin stimulation. As shown in Fig. 3, leptin stimulation significantly increased ERK gene expression. Additionally, the activation of ERK, assessed by measuring the levels of phospho-specific ERK, was found to increase in response to leptin. However, we did not detect dose-dependent activation of ERK by leptin stimulation (Fig. 3).
Effects of leptin on the activation of MEK and ELK
We next examined the effect of leptin on the activation of MEK, which is an upstream activator of MAPK. Phospho-specific MEK, an indication of MEK activation, occurred in response to leptin stimulation. Similar to ERK activation, we could not find a dose-dependent activation of MEK by leptin stimulation; however, there was no significant difference in total MEK protein expression among the groups (Fig. 4). In addition, we determined whether activation of the MEK-MAPK pathway could induce a parallel increase in the activity of a MAPK target transcription factor, ELK. As shown in Fig. 4, ELK activation measured by the level of phospho-specific ELK was also observed after leptin stimulation. However, no change in total ELK protein expression was observed based on the concentration of leptin.
Effects of leptin on sequential activation of the MAPK pathway
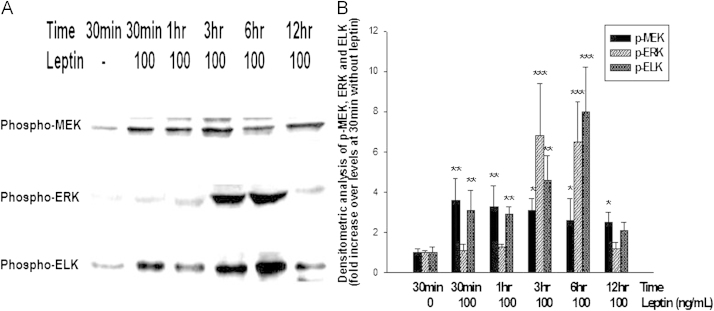
Finally, we determined whether leptin stimulation induced a sequential activation of the MAPK pathway, we examined the activation of MEK, ERK, and ELK in response to leptin at a final concentration of 100 ng/mL, and harvested after 30 minutes, 1 hour, 3 hours, 6 hours, and 12 hours. As shown in Fig. 5, the activation of MEK was found to rapidly increase in response to leptin after 30 min, whereas maximal activation of ERK and ELK was observed at the 3- and 6-hour intervals, respectively.
Discussion
Obesity significantly contributes to the development of atherosclerosis and consequent cardiovascular disease. One plausible, recently proposed mechanism is that factors known as adipocytokines, produced by adipose tissue, directly impact the atherogenic environment of the vessel wall by regulating gene expression and function in endothelial, arterial smooth muscle, and macrophage cells. The reduction of visceral fat mass leads to amelioration of these risk factors and potentially prevents cardiovascular events [15]. Although multiple molecular mechanisms contribute to the development of obesity-related complications, recent data suggest that inflammation, and especially monocytes/macrophages, is a central axis in the pathophysiology of many obesity-related disorders [16].
In the present study, we found that plasma leptin levels were significantly higher in type 2 diabetic participants than in healthy controls irrespective of the stage of diabetic nephropathy. Considering that the study groups were not significantly different in body weight and BMI, these results suggest that diabetes is the main cause of elevated leptin concentrations in diabetic participants, rather than obesity. Interestingly, plasma levels of leptin had an increased tendency according to the status of diabetic nephropathy, and we detected a positive correlation with urinary albumin excretion.
Considering that albuminuria is a surrogate marker of vascular dysfunction, these results suggest that plasma leptin levels are elevated in participants with a high risk of cardiovascular disease; however, we did not detect significant differences in plasma leptin levels between the normal and microalbuminuric participants. Furthermore, plasma leptin levels were negatively correlated to creatinine clearance, which is in agreement with previous reports [17]. Although there is a possibility that a decreased clearance of leptin in a low glomerular filtration rate (GFR) state may contribute to increased plasma leptin levels, or that increased plasma leptin levels may induce renal injury, the mechanism by which plasma leptin levels increase with decreased GFR remains unclear. It is also unclear whether circulating leptin levels are influenced by renal function, because there are not enough data on circulating leptin levels according to GFR in participants with chronic kidney disease (CKD). In the present study, we included participants with early stages of CKD in type 2 diabetic nephropathy; however, most participants did not exhibit stage 4 or 5 CKD.
Plasma MCP-1 levels were similarly elevated in diabetic participants and controls. Plasma MCP-1 levels were the only independent risk factor for plasma leptin levels after adjustments for all parameters. Collectively, these findings suggest that plasma concentrations of leptin and MCP-1 are elevated under diabetic conditions.
Leptin is a peptide hormone synthesized by adipose tissue and plays a role in the regulation of food intake and energy expenditure. Elevated leptin levels, which often coexist with diabetes and metabolic syndrome, have been considered as an independent risk factor for the progression of cardiovascular diseases, including atherosclerosis and hypertension. The classic effect of leptin on food intake and energy expenditure is mediated by the long form of leptin receptor, which is mainly expressed in the hypothalamus, but the roles of short forms of leptin receptors that are widely expressed in the body are not yet clear [18]. Growing evidence shows that leptin plays additional roles in the regulation of the hypothalamic-pituitary-peripheral axis, insulin resistance, and immunity [19], [20]; however, the role of leptin in the pathogenesis of atherosclerosis remains unclear. VSMCs play a vital role in arterial intimal thickening and vascular remodeling. Bohlen and colleagues [21] reported that a short form of leptin receptor, leptin receptor (OB-R), was mainly expressed compared to the long isoform, in cultured human VSMCs.
We further investigated the effect of leptin on MCP-1 synthesis to define the molecular mechanism and direct the effects of leptin in cultured VSMCs. We observed that MCP-1 synthesis was significantly increased by leptin stimuli. In addition, we observed that leptin-induced MCP-1 synthesis was significantly inhibited by a MEK inhibitor. This result suggests that leptin-induced MCP-1 production might be mediated by the MEK-MAPK (ERK) pathway. Furthermore, we observed that leptin stimulation sequentially activated upstream MEK, MAPK, and downstream ELK signaling pathways.
Increasing evidence demonstrates that circulating leptin levels may contribute to the pathogenesis of atherosclerosis. In a relatively large cohort study, high leptin levels were an independent risk factor for coronary heart disease [22]. Additionally, leptin levels were higher in type 2 diabetes participants with coronary artery calcification measured with electron beam tomography [23]. In another study, high leptin levels were also a predictor of angiographic coronary artery atherosclerosis [24]. Furthermore, exogenous leptin injection promoted atherosclerosis in apolipoprotein E-deficient mice [25]; however, several human studies showed contradictory results that low leptin levels were associated with low cardiovascular mortality, and leptin levels were significantly lower in participants with coronary artery stenosis than in control participants [26], [27]. It should be noted that the study sampled used in the previous report is different from that used in our study.
There is not enough evidence for the direct effects of leptin on VSMC physiology. Previous studies have observed that leptin induces a proliferation and migration of VSMCs that is important in the pathogenesis of atherosclerosis [3]. In other studies, leptin stimulated the proliferation of VSMCs by promoting transition from the G1 to S phase [3], and through a protein kinase C-dependent activation of Nicotinamide Adenine Dinucleotide Phospate Reductase (NAD(P)H) oxidase [28]. In this study, we observed for the first time that leptin stimulates MCP-1 synthesis via the MEK-MAPK pathway. These results are in agreement with those of recent reports that leptin increases MCP-1 production in hepatocytes, and that leptin-induced MAPK activation may be associated with VSMC proliferation and the progression of atherosclerosis [28], [29], [30], [31].
In conclusion, this study suggests a new physiologic role of leptin in atherosclerosis in diabetic participants. Plasma levels of leptin are markedly increased in diabetic participants irrespective of the stage of nephropathy. Plasma leptin levels were independently associated with plasma levels of MCP-1. In addition, leptin increased MCP-1 production and this stimulating effect of leptin on MCP-1 expression was reversed by MEK inhibitor PD98059. Overall, these findings suggest that activation of leptin synthesis in a diabetic environment may promote MCP-1 activation via the MAPK pathway in VSMCs, and possibly contributes to the acceleration of atherosclerosis.


 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









