Fibrillary glomerulonephritis combined with chronic inflammatory demyelinating polyneuropathy
Article information
Abstract
A 58-yr-old man presented with leg edema and subacute weakness of his bilateral lower extremities. Urinary and serum immunoelectrophoresis revealed the presence of lambda-type Bence Jones proteins. He was ultimately diagnosed with monoclonal gammopathy of undetermined significance (MGUS). A renal biopsy specimen showed fibrillary glomerulonephritis (FGN), which was randomly arranged as 12–20 m nonbranching fibrils in the basement membranes. Immunofluorescence studies were negative for immunoglobulin (Ig)G, IgM, IgA, C3, and kappa light chains in the capillary walls and mesangial areas. A Congo red stain for amyloid was negative. Electromyography and nerve conduction velocity examinations results were compatible with the presence of demyelinating polyneuropathy. This case showed a rare combination of FGN, without Ig deposition, and MGUS combined with chronic inflammatory demyelinating polyneuropathy (CIDP).
Introduction
Fibrillary glomerulonephritis (FGN) is an idiopathic glomerular disease characterized by randomly arranged, Congo red-negative fibrils, 16–24 nm in diameter [1,2]. Glomerular deposits, predominantly composed of polyclonal immunoglobulin (Ig)G and C3, are also commonly observed [3,4]. This disorder is found in <1% of patients who undergo renal biopsy, and usually presents with renal insufficiency, nephrotic range proteinuria, and microhematuria [2].
FGN is sometimes associated with autoimmune diseases or lymphoproliferative disorders, and is associated with serum or urine monoclonal gammopathy in 15% of patients [4]. Monoclonal gammopathy of undetermined significance (MGUS) can also be associated with peripheral neuropathy [5]. Neuropathies associated with MGUS include the full spectrum of neuropathic disorders, which can be either axonal or demyelinating [5]. To our knowledge, this is the first case exhibiting the pathological characteristics of FGN, without Ig deposits, combined with chronic inflammatory demyelinating polyneuropathy (CIDP).
Case report
A 58-yr-old man was admitted to our hospital because of leg edema, which had appeared >2 months earlier, and bilateral lower extremity weakness. He had a 10-year history of diabetes mellitus and hypertension.
A laboratory urinalysis revealed 3+ protein and rare red and white blood cells. A 24-hour urine collection revealed the excretion of 4.7 g of protein. Serum chemistry disclosed the following: blood urea nitrogen, 38 mg/dL; creatinine, 1.73 mg/dL; total protein, 6.8 g/dL; albumin, 4.2 g/dL; calcium, 8.9 mg/dL; and total cholesterol, 91 mg/dL. The patient’s liver function test results were within normal limits, but serum and urine electrophoresis and immunofixation results disclosed the presence of a monoclonal Ig light chain (lambda-type). His serum IgG, IgA, and IgM levels were 946 mg/dL, 114 mg/dL, and 18.7 mg/dL, respectively; his urinary level of b2 microglobulin was >20,000 µg/L. A cerebrospinal fluid study revealed results within normal limits. Serological test results for underlying causes of glomerulonephritis were normal, and a bone survey failed to reveal any abnormalities. A bone marrow aspiration test revealed mild plasmacytosis (7.0% of the total cells). Based on these findings, the patient was diagnosed with MGUS.
The results of a renal biopsy showed variable mesangial widening of the glomeruli, with focal capillary wall thickening (Fig. 1A). A Masson-trichrome stain showed mesangial and capillary accumulations that appeared as pale blue foci. Global sclerotic glomeruli and extracapillary crescent formations of glomerular tufts were not observed, but the renal vessels revealed subintimal thickening with wall fibrosis. Mild tubular atrophy or loss was also noted, along with chronic mononuclear inflammatory infiltrates that were sparsely present in the interstitium. There was no evidence of diabetic nephropathy, such as Kimmelstiel-Wilson nodules. An immunofluorescent examination did not reveal IgG, IgA, IgM, or C3 (granular glomerular stain) in the mesangial spaces or along the capillary walls. A stain for kappa and lambda light chains, in the glomeruli, was negative. A Congo red stain for amyloid was also negative.
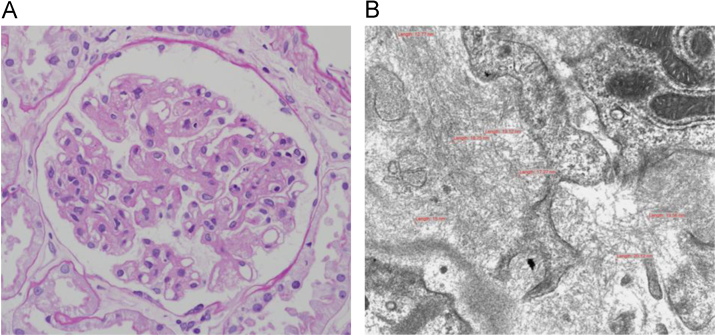
Renal biopsyfindings. (A) A light micrograph shows variable glomerular mesangial widening and focal capillary wall thickening. (B) Electron microscopy shows fibrillary glomerular deposits composed of randomly arranged, straight, nonbranching fibrils, 12–20 nm in diameter. These fibrils are seen disrupting the glomerular basement membrane and in the mesangium.
Electron microscopy of the biopsy specimen revealed fibrillary glomerular deposits of randomly arranged, straight, nonbranching fibrils, ranging in diameter from 12 nm to 20 nm, disrupting the glomerular basement membranes and on the mesangium (Fig. 1B); podocyte foot process effacement was also observed. The capillary walls showed focal wrinkling and irregular thickening.
We performed electromyography and checked nerve conduction velocity in the patient’s upper and lower extremities. These tests revealed demyelinating sensorimotor polyneuropathy, consistent with CIDP (Table 1).

Electromyography and nerve conduction velocity results
Ultimately, the patient was diagnosed with FGN, without Ig deposition, and CIDP associated with MGUS. We started treatment with prednisolone (60 mg/d) for 6 weeks and intravenous Ig (2.5 g/d) for 5 days. After the treatment, the patient experienced worse upper and lower extremities and had bad serum chemistry (blood urea nitrogen, 73 mg/dL; creatinine, 2.17 mg/dL) diagnosed worse than before the treatment (Fig. 2). He was then transferred to another hospital at the request of the patient.
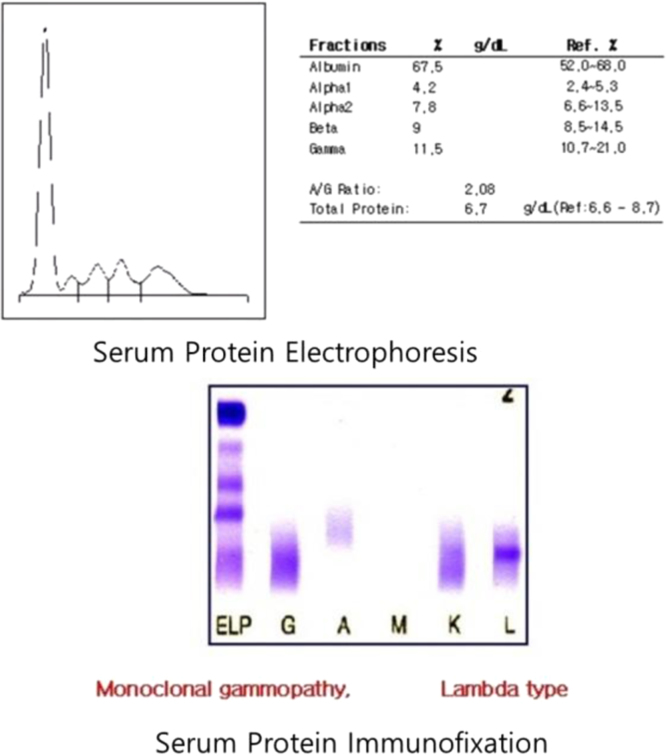
Serum protein electrophoresis and immunofixation.
Discussion
This patient is the first reported case of FGN, without Ig deposition, combined with CIDP and lambda-type light chain-MGUS. There is considerable debate about the relationship of FGN to immunotactoid glomerulonephritis, but these diseases have different pathological characteristics [1–4]. FGN is a distinctive idiopathic glomerular disease characterized by randomly arranged, Congo red-negative fibrils, 16–24 nm in diameter [1–4]. By contrast, immunotactoid glomerulonephritis is an entity with larger microtubular deposits, usually >30 nm in diameter, which are often hollow and arranged in parallel or stacked arrays [3,4]. The fibrils usually stain positive for IgG and C3, with more variable and weaker positivity for other Igs [1–3]. Nasr et al [6] reported 66 cases of FGN, which showed 100% glomerular positivity for IgG, 28% positivity for IgA, and 47% positivity for IgM. FGN is sometimes associated with MGUS, which is characterized by the presence of a monoclonal gammopathy without end-organ damage [4]. In the present case, immunofluorescence studies were negative for IgG, IgM, IgA, C3, kappa, and lambda light chains in the capillary walls, mesangial areas, and glomeruli. Churg and Venkataseshan [7] also reported some cases of FGN without Ig deposition.
The pathogenesis of FGN remains unknown [3], but several studies have provided evidence that the fibrils, in this disorder, are composed of Ig [4]. Therefore, Igs are believed to play an important role in fibrillogenesis [4]. By contrast, Churg and Venkataseshan [7] suggested that FGN is not a homogeneous entity and has a variety of precursors, including Igs and other, as yet unidentified, substances. It is the case with monoclonal gammopathy and Bence-Jones proteinuria over 20,000 µg/L. If there are no depositions of Ig in renal biopsy, we consider possible causes: (1) there is a lack of light chain deposit despite FGN with monoclonal gammopathy; and (2) the only conformational changed light chain and Ig were deposited. Currently, there is no way to distinguish these causes. Further studies may be required [7].
MGUS can be associated with peripheral neuropathy, but it is often not clear whether the pathogenesis of the neuropathy involves paraproteins or merely represents a coincidental association [5]. In patients with neuropathy, the most commonly involved monoclonal protein is the IgM class (48%), followed by IgG (37%), and IgA (15%) [5]. In some cases, evidence suggests that the M protein cross-reacts with a neural antigen such as myelin-associated glycoprotein (a glycoprotein found in the myelin sheath of the central and peripheral nerves), sulfated glucuronyl paragloboside, or sulfatide, resulting in complement activation and nerve damage [5,8]. In the present case, only light chain proteins were found in conjunction with CIDP.
As highlighted by this case, electron microscopy findings are important because some FGN patient biopsy samples may not stain in an immunofluorescence study. Furthermore, if neurologic symptoms are accompanied by Bence-Jones proteinuria, the patient should be assessed for paraproteinemic demyelinating neuropathy.
Conflict of interest
All authors declare no conflict of interest.