


Introduction
End-stage renal disease (ESRD) due to diabetes has been estimated to be 30–47% of all incident cases worldwide [1]. Disparities in the incidence of ESRD from diabetes among ethnic groups have existed for many years, but the magnitude has been increasing. Diabetic nephropathy (DN) develops along with generalized microvascular disease, most often concomitant with macrovascular disease including cardiovascular, cerebrovascular, and peripheral arterial diseases. Patients with DN have a higher risk of mortality, mostly from cardiovascular complications, than diabetic patients without nephropathy [2].
Risk factors
The epidemiology of DN has been best studied in patients with type 1 diabetes, because the time of clinical onset is usually known. The onset of overt nephropathy in type 1 diabetes is typically between 10 and 15 years after the onset of the disease. Both environmental and genetic factors have been postulated as DN risk factors. Poor glycemic control, long duration of diabetes, insulin resistance, high blood pressure (BP), advanced age, smoking, race, and genetic predisposition are the main risk factors for the development of DN. Many genes have been implicated as conferring DN risk [3].
DN is a classic complex trait, whose development in a given individual likely reflects contributions from multiple genes whose expression is modulated by environmental factors. Numerous genetic strategies have been used to identify common disease risk loci and genes, including candidate gene analyses, family-based linkage analysis and transmission disequilibrium testing, population-based admixture mapping, and genome-wide association studies (GWAS) [4]. Candidate gene-based association has been the most common approach used to identify susceptibility genes for DN. Genes encoding for angiotensin-converting enzyme, angiotensin II (Ang II) receptor, various aspects of glucose metabolism, lipid metabolism (apolipoprotein E gene polymorphism), extracellular matrix, and inflammatory cytokines have been selected to test for an association with DN based on the pathogenesis of disease [5], [6]. Genome-wide linkage analysis facilitates the identification of previously unsuspected genes as risk factors. It is most powerful when the frequency of the polymorphism is low but the effect size is high. An early family-based genome-wide linkage analysis from the Family Investigation of Nephropathy and Diabetes (FIND) research group identified chromosomal loci for susceptibility genes, including 1q, 7q, and 18q linked to estimated glomerular filtration rate (GFR), in a multiethnic collection of families ascertained by a proband with type 2 diabetes and DN [7]. Using linkage analysis and the identification of positional candidate genes under the linkage peaks, others identified polymorphisms in the carnosinase 1 gene on chromosome 18q [8], the adiponectin gene on 3q [9], and the engulfment and cell motility (ELMO1) gene on 7p [10] as DN risk genes. GWAS have greater power than linkage analysis to identify polymorphisms when the gene effect size is low, but the frequency of the polymorphism in the population is high. GWAS identified several novel risk loci including—but not limited to—SLC12A3
[11]; ELMO1
[12]; 4.1 protein ezrin, radixin, moesin domain containing 3 (FRMD3) [13]; and SAM and SH3 domain containing 1 (SASH1) gene [14]. Collaboration among many genetic research groups around the world with thousands of samples and clinical databases continue to seek replicable genetic polymorphisms that confer DN risk.
Reflecting an appreciation for genetic–environmental interactions in DN development, an emerging science has evolved defining contributions of epigenetics to the development of DN. A growing number of pathogenetically important microRNAs (miRs) have been identified in DN [15], representing opportunities for risk assessment and therapeutic targeting.
Clinical staging
Renal disease in diabetic patients had been clinically characterized by increasing rates of urinary albumin excretion and decreasing renal function, with at-risk patients marching through the stages of normoalbuminuria, microalbuminuria, overt proteinuria, and finally ESRD. However, with treatment, not only can progression be slowed, but there is also some plasticity in this staging, and regression from a more severe to a less severe stage can sometimes be achieved. In the susceptible, normoalbuminuria progresses to microalbuminuria, macroalbuminuria, and eventually to ESRD. Persistent albumin excretion between 30 mg/d and 300 mg/d is defined as microalbuminuria. Regression from microalbuminuria to normoalbuminuria occurs spontaneously in a substantial proportion of diabetic patients [16]. Nevertheless, patients with persistent microalbuminuria are at high risk of progressing to overt nephropathy and developing cardiovascular disease [17]. Albuminuria in excess of 300 mg/d represents overt nephropathy. Once overt proteinuria occurs, there is concomitant loss of GFR in both type 1 and type 2 diabetes. Hypertension exacerbates GFR loss. Historically, studies dealing with the natural history of DN demonstrated a relentless, often linear but highly variable rate of decline in GFR ranging from 2 mL/min/y to 20 mL/min/y (mean 12 mL/min/y) [18]. However, the rate of decline may be substantially less with tight BP and blood glucose control. In a recent study, the rate of GFR decline ranged from 0 mL/min/y to 4 mL/min/y [19]. Thus, many patients who are well treated may achieve stable renal function for long periods.
Pathogenesis
Hyperglycemia-induced metabolic and hemodynamic stimuli are mediators of kidney injury [20], [21]. These activate inflammatory, pro-oxidant, ischemic, and fibrotic pathways leading to mesangial matrix accumulation; podocyte effacement and loss; glomerular basement membrane (GBM) thickening; endothelial dysfunction; tubular atrophy, fibrosis, and dropout; tubulointerstitial inflammation, and renal arteriolar hyalinosis [20].
Hemodynamic factors
The hemodynamic factors contributing to DN involve increased systemic and intraglomerular pressure and activation of various vasoactive hormones, including the intrarenal renin–angiotensin–aldosterone system (RAAS), nitric oxide, vascular endothelial growth factor (VEGF), and endothelin. Hemodynamic changes play an important role, being present early in the disease, exacerbating albumin passage across glomerular capillaries, and contributing to mesangial matrix expansion, podocyte injury, and nephron loss [22].
Metabolic factors
Hyperglycemia accelerates the development of renal disease by increasing intracellular glucose availability. The facilitative glucose transporter, GLUT1 mediates mesangial cell glucose flux, which leads to the activation of signaling cascades favoring glomerulosclerosis, including pathways mediated by transforming growth factor β (TGF-β), advanced glycosylation end products (AGEs), protein kinase C, and various cytokines and growth factors [23]. In addition, decreased phosphorylated p38 (pp38) mitogen-activated protein kinase (MAPK) after chronic glycemic stress may contribute to podocyte cytoskeletal alterations and albuminuria [24].
In chronic hyperglycemia, glucose combines with free amino groups on circulating or tissue proteins. This nonenzymatic process initially forms reversible early glycosylation products and later irreversible AGEs. AGEs activate specific receptors, inducing cellular dysfunction and injury. AGEs contribute to the accumulation of glomerular extracellular matrix proteins, associated with a concomitant depression in collagenase activity. An AGE-related functional defect in the permselective properties of the podocyte slit membrane may contribute to the development of albuminuria [25].
Oxidative stress/inflammation
Traditionally, hyperglycemia-induced overproduction of reactive oxygen species (ROS) in diabetes has been implicated in the pathogenesis of diabetic complications [26]. Some have recently challenged the hypothesis that increased cellular superoxide production underlies DN. In experimental models, Dugan et al [27] show that DN may well be characterized by low mitochondrial superoxide production, and that increased mitochondrial superoxide production may attenuate DN. Therefore, in addition to glycemic and BP control, restoration of mitochondrial structure, function, and signaling may be novel ways to improve DN and prevent the decline in organ function.
Metabolic pathways are the major mediators of DN. They promulgate activation of the immune system and chronic inflammation. Several studies suggest that the small increment in monocytes/macrophages observed in glomeruli contribute significantly to the evolution of DN. Intrinsic renal cells, including mesangial, glomerular endothelial, dendritic, and renal tubular cells, are able to produce inflammatory cytokines and growth factors, mainly VEGF, TGF-β, interleukin 1 (IL-1), IL-6, and IL-18, as well as tumor necrosis factor α (TNF-α), which have all been implicated in DN progression [28].
Matrix protein accumulation is a major determinant of progressive renal injury in DN. It can result from increased synthesis and/or decreased degradation of matrix proteins [29]. Recently discovered noncoding RNAs such as miRs usually act as inhibitors of mRNA translation. A growing number of miRs have been implicated in the development of DN, including—but not limited to—192, 21, 29c, 93, 141, 216a, 377, and the 200 family [30], [31]. These have been implicated in mediating inflammation and fibrosis in DN [32], [33]. miRs are undergoing intense scrutiny for their value in elucidating the pathogenesis of DN and for their potential as therapeutic targets.
Diagnostic criteria
The following recommendations are based on the clinical practice guidelines for diabetic kidney disease outlined by the Kidney Disease Outcomes Quality Initiative (KDOQI), which were last updated in 2012 [34]. Screening for albuminuria should begin at 5 years’ diabetes duration in patients with type 1 diabetes and at the time of diagnosis in patients with type 2 diabetes. The preferred screening test is a urine albumin/creatinine ratio with a first-morning void spot collection. If microalbuminuria is present, the test should be confirmed for persistence with two of three positive repeat measurements within 6 months. DN is likely in patients with diabetes, persistent microalbuminuria or overt proteinuria, a duration of diabetes of at least 10 years and/or diabetic retinopathy, and in the absence of other clinical and/or historical factors suggesting additional or alternative causes for abnormal albuminuria. A renal biopsy may be necessary to confirm the clinical diagnosis if atypical features are present, including the appearance of nephropathy earlier than anticipated, the presence of a nephritic sediment, a more rapid loss of renal function than anticipated, or the presence of serological abnormalities obtained during screening. The presence of retinopathy evaluated through a routine ophthalmologic exam was thought to predict nephropathy. Retinopathy correlates well with overt nephropathy and declining GFR <30–60 mL/min/1.73 m2. However, the association is not as strong in early diabetes [35] and is less predictive in type 2 diabetes than type 1 diabetes.
Renal pathology
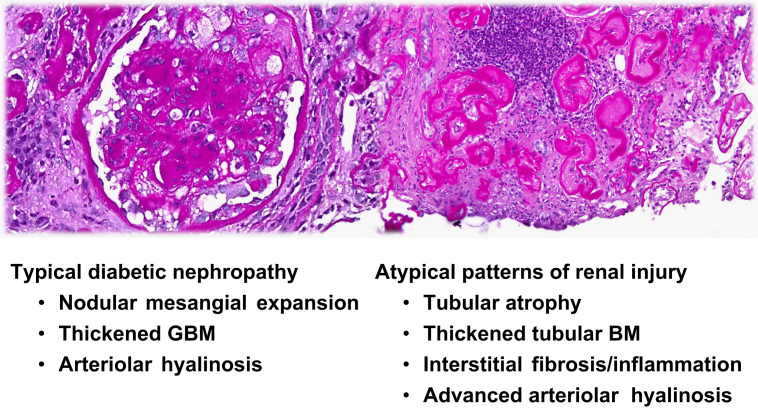
The histopathological lesions of DN have recently been classified (Table 1) [36]. Renal pathological changes are present in patients with long-standing diabetes prior to the onset of microalbuminuria [37]. The characteristic light microscopic features of DN comprise three major lesions: thickened GBM and tubular basement membranes, diffuse mesangial expansion, and hyalinosis of afferent and efferent arterioles (Fig. 1). In the new classification, Class I consists of electron microscopy-confirmed thickening of the GBM, adjusted for gender and age. Class II consists of mild (IIA) to severe (IIB) mesangial expansion. GBM thickening and mesangial matrix accumulation are the first changes that may occur at 2–5 years of diabetes. The degree of mesangial expansion correlates inversely within the capillary filtration surface area, which contributes to the progression from hyperfiltration to reduced GFR [38]. Class III consists of nodular glomerulosclerosis, a lesion first described by Kimmelstiel and Wilson in 1936. Finally, Class IV consists of advanced DN, comprising >50% global glomerulosclerosis along with additional lesions of Classes I, II, or III. Tubulointerstitial inflammation and atrophy and vascular lesions are scored separately in scales of 0–3 or 0–2. Arteriolar hyalinosis, arteriosclerosis, glomerular capillary subendothelial hyaline (hyaline caps), and capsular drops along the epithelial parietal surface of the Bowman capsule (e.g., the so-called exudative lesions of DN) may also be present. The classification uses electron microscopy only to measure GBM thickness; it does not include podocyte changes, nor has it been tested for its predictive value for clinical or research utility. Numerous relationships between tubulointerstitial change and functional outcomes have been reported. Interstitial fibrosis is often proportional to tubular atrophy and a strong predictor of the rate of progression from moderate to severe reduction in GFR [39], [40]. Urinary biomarker data in human beings support the view that tubule injury contributes in a primary way, rather than in a secondary manner, to the development of early DN [41].
Therapeutic interventions
General measures for prevention and treatment of DN, and protection against cardiovascular morbidity and mortality include rigorous BP control with RAAS inhibitors (RAASi), glycemic control, treatment of dyslipidemia, as well as diet and lifestyle modifications, including physical activity, appropriate weight reduction, and smoking cessation (Table 2).
Glycemic control
Glycemic control prevents and improves microvascular complications [42], [43]. The efficacy of glycemic control as a renoprotective strategy depends in part on the stage at which it is begun and the degree of normalization of glucose metabolism. Glycemic control can partially reverse early glomerular hyperfiltration and new-onset microalbuminuria [43], [44], [45]. Glycemic control can also stabilize and/or retard progression in diabetic patients with overt nephropathy [46]. However, few studies address intensive glycemic control in patients with advanced DN, in whom it may be difficult to show a benefit.
Type 1 diabetes
The Diabetes Control and Complications Trial (DCCT) compared the effects of intensive glucose control with conventional treatment on the development and progression of long-term complications of type 1 diabetes. During a 9-year period, patients receiving intensive therapy [mean hemoglobin A1c (HbA1c) 7%] had a 35–45% lower risk for development of microalbuminuria compared with the control group (mean HbA1c 9%) [45]. More recently, the Epidemiology of Diabetes Interventions and Complications (EDIC) trial data indicated that the long-term risk of an impaired GFR was 50% lower in patients treated for an average of 12 years with the DCCT-intensive glucose control regimen compared to those treated with conventional therapy. This effect was not evident until more than 10 years after randomization, beyond the period of the DCCT treatment intervention [47]. This indicates that early and long-term control of hyperglycemia significantly ameliorates DN and that the beneficial effect persists even when glucose control is relaxed.
Moreover, the benefits of glycemic control after pancreas transplantation in patients with type 1 diabetes were observed in that mesangial matrix volume, thickening of glomerular and tubular basement membranes, and nodular glomerular lesions were significantly reduced or returned to normal. However, histologic remodeling was a slow process, taking approximately 10 years after transplantation [48], [49].
Type 2 diabetes
The Kumamoto study reported a 60% reduction in microalbuminuria in young type 2 diabetic patients achieving an HbA1c of 7.0% [50]. In the United Kingdom Prospective Diabetes Study (UKPDS) trial, newly diagnosed patients with type 2 diabetes were randomly assigned to intensive management (HbA1c 7.0%) with a sulfonylurea or insulin or to conventional management (HbA1c 7.9%) with diet alone. After 9 years of treatment, tight glycemic control reduced the incidence of microalbuminuria by 24% [43]. Moreover, after termination of the UKPDS study, patients with type 2 diabetes randomized to the lower HbA1c target continued to experience risk reduction for myocardial infarction and death from any cause up to 10 years after the original randomized assignment, in some cases despite higher subsequent HbA1c after the study’s conclusion [51]. This phenomenon of ongoing beneficial effects on diabetic complications after a period of improved glycemic control even if followed by a return to less intensive metabolic control has been described as a “legacy effect” by the UKPDS investigators. This observation underlies the importance of early glycemic control before complications develop.
More recent studies, including the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial, Action in Diabetes and Vascular Disease, Perindopril and Indapamide Controlled Evaluation (ADVANCE) trial, and the Veterans Affairs Diabetes Trial (VADT), which targeted lower HbA1c goals (<6–6.5%), failed to show that extremely tight glycemic control protects against macrovascular complications in patients with relatively advanced age and long duration of type 2 diabetes. Although some benefit for albuminuria may have been achieved [52], [53], [54], in the ACCORD trial, very tight glycemic control was associated with a 22% increase in mortality from any cause [52]. Thus, the effects of intensive glycemic control on the prevention of macrovascular complications are less certain, particularly in patients with long disease duration. A recent systematic review revealed that intensive glucose control reduced the risk for microalbuminuria and macroalbuminuria, but evidence is lacking that intensive glycemic control reduces the risk for doubling of creatinine, ESRD, or death [55].
Decreasing insulin requirements and frequent hypoglycemia occur in advanced chronic kidney disease (CKD) and in patients on dialysis. Impaired insulin sensitivity and deficient gluconeogenesis—along with malnutrition, chronic inflammation, deficient catecholamine release, and impaired renal insulin degradation and clearance—can contribute to low blood glucose levels in patients with CKD and ESRD [56], [57], [58]. Therefore, aggressive glycemic control cannot be routinely recommended for all DN patients for the purpose of reducing mortality risk. Glucose-lowering treatment must be individualized in DN patients.
Both KDOQI and Kidney Disease Improving Global Outcomes (KDIGO) recommended a target HbA1c of <7% or ~7%, respectively, regardless of the presence or absence of CKD, a goal that is in line with diabetes management in the general population [59]. However, these recommendations are not strongly evidence-based, because few studies address the benefits and risks of intensive glycemic control in late stages of CKD or ESRD [60], [61]. Patients likely to benefit the most from tight glycemic control include those with short diabetes duration, long life expectancy, and no significant cardiovascular disease. Less-stringent HbA1c goals (such as <8%) may be appropriate for patients with a higher risk of hypoglycemia, difficult glycemic control, those with hypoglycemia unawareness, limited life expectancy, extensive comorbid conditions, and/or advanced microvascular and macrovascular complications, including advanced CKD [61] (Table 3). Many hypoglycemic agents are renally excreted, requiring dosage adjustment in CKD (Table 4).
Glycemic monitoring in CKD
Currently, HbA1c remains the most accurate method to assess chronic glycemic control [43]. HbA1c underestimates glycemic control in advanced CKD and ESRD because of shorter erythrocyte life span, iron deficiency anemia, recent transfusion, and erythropoietin treatment [62]. Despite the limitations of HbA1c in advanced CKD and ESRD, HbA1c is still considered a reasonable measure of chronic glycemic control in this group. Glycated albumin and fructosamine have also been tested [63], [64], although each of these is also beset by confounding factors. Thus, for patients prone to glycemic variability (especially type 1 diabetes patients, or type 2 diabetes patients with severe insulin deficiency), glycemic control is probably still best assessed by a combination of the results of self-monitoring of blood glucose testing and the HbA1c [61].
BP control
BP target
In both type 1 and type 2 diabetic patients, early treatment of hypertension is critical for DN prevention and treatment. However, the optimal lower limit for BP control in DN remains unclear. Major guidelines published prior to the ACCORD BP trial suggested that the target BP in diabetic patients should be <130/80 mmHg [59]. However, ACCORD challenged this BP target. Among diabetic patients with high cardiovascular risk randomized to a goal systolic BP <120 mmHg versus standard therapy with a goal <140 mmHg, there was no difference in the risks of composite major cardiovascular events; but increased rates of hyperkalemia and renal dysfunction were observed when targeting a systolic BP of <120 mmHg [65]. In a secondary analysis of the Irbesartan Diabetic Nephropathy Trial (IDNT), progressive lowering of systolic BP to 120 mmHg was associated with improved renal and patient survival, an effect independent of baseline renal function [66]. Thus, given the lack of strong evidence of benefit from reducing systolic BP to below 130 mmHg, some may target <140/90 mmHg as a BP goal for diabetic patients. A target of 130/80 mmHg or less can be pursued in patients with DN or CKD, younger patients, patients who tolerate their antihypertensives well, and patients at high risk for stroke. The KDIGO clinical practice guideline for the management of BP in CKD recommended thresholds to initiate treatment to lower BP of 130/80 mmHg and 140/90 mmHg for diabetic patients with and without urine albumin excretion >30 mg/d, respectively [67]. In addition, KDIGO recommended individualized BP targets and agents according to age, coexistent cardiovascular disease and other comorbidities, risk of progression of CKD, presence or absence of retinopathy, and tolerance of treatment.
RAASi
The RAAS has key regulatory functions for BP and sodium homeostasis. In particular, Ang II, the main effector of the RAAS, enhances the vascular tone of both afferent and efferent glomerular arterioles by interacting with angiotensin type 1 and type 2 receptors (AT1, AT2), thereby modulating intraglomerular pressure. Besides the hemodynamic effects, activation of AT1 receptors triggers the expression and release of a range of proinflammatory and profibrotic mediators implicated in DN progression [68]. In hypertensive diabetic patients, angiotensin-converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARBs) are effective first-line antihypertensive agents and reduce DN disease progression [69].
RAASi in type 1 diabetes
In type 1 diabetes with persistent microalbuminuria and overt nephropathy, several studies showed that ACEi lower albuminuria and decrease the risk of renal progression [70], [71]. The first study was published more than 20 years ago. In the landmark randomized, controlled trial comparing captopril with a placebo in patients with type 1 diabetes in whom urinary protein excretion was >500 mg/d and the serum creatinine concentration was ≤2.5 mg/dL, captopril treatment attenuated renal functional decline and reduced the risk of the composite end point of death, dialysis, and doubling of serum creatinine [72]. Subsequent meta-analyses have since confirmed this finding [73]. There are no large long-term clinical trials to demonstrate the efficacy of ARBs in type 1 diabetes with DN. Nevertheless, based on the shared properties of ACEi and ARBs, there is reason to believe that both are effective in the treatment of type 1 DN.
In patients with normotensive and normoalbuminuric type 1 diabetes, most—but not all—clinical trials show no benefit of RAASi on nephropathy progression [74], [75], [76]. The KDOQI 2012 Diabetes Guideline recommended not using an ACEi or ARB for the primary prevention of DN in normotensive normoalbuminuric patients with diabetes [34].
RAASi in type 2 diabetes
In hypertensive normoalbuminuric type 2 diabetic patients, olmesartan reduced the incidence of microalbuminuria from 9.8% to 8.2% even though BP control in treatment and control groups were excellent according to current standards. Of concern was a higher rate of fatal cardiovascular events with this ARB among patients known to have preexisting coronary heart disease, especially those with lower BP [77]. The Bergamo Nephrologic Diabetes Complications Trial (BENEDICT), which randomized hypertensive normoalbuminuric type 2 diabetic patients to placebo, verapamil, trandolapril, or a combination of verapamil plus trandolapril, showed less progression to microalbuminuria in patients receiving trandolapril either alone or with verapamil [74]. Overall, the risks appear to exceed the benefit in using RAASi prophylactically to prevent microalbuminuria.
In the stage of microalbuminuria, the Irbesartan in Patients with Type 2 Diabetes Microalbuminuria (IRMA 2) study showed that the ARB reduced progression to overt nephropathy by 70% in hypertensive type 2 diabetic patients during a 2-year follow-up period [78]. In the MicroAlbuminuria Reduction With VALsartan (MARVAL) study, the ARB produced a greater reduction in albuminuria compared with amlodipine with the same degree of BP reduction, suggesting that the antiproteinuric effect of the ARB is BP-independent [79]. RAASi are recommended to slow the progression from microalbuminuria to overt proteinuria.
Two landmark trials now more than a decade old showed clear benefit for ARBs in the treatment of type 2 diabetes with overt nephropathy. In the IDNT study, 1,715 hypertensive patients with nephropathy due to type 2 diabetes were randomly treated to irbesartan, amlodipine, or placebo. Treatment with irbesartan showed a relative risk reduction of the primary composite end point (doubling of the plasma creatinine, development of ESRD, or death from any cause) in the irbesartan group [80]. In the Reduction of End point in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial, 1,513 patients with DN were randomly assigned to losartan or placebo, both in addition to conventional antihypertensive therapy. At 3.4 years, losartan reduced the same composite end point by 16%. Losartan reduced the incidence of serum creatinine doubling by 25% and the risk of ESRD by 28% [81]. Both studies showed that the benefit of ARBs exceeded that attributable to changes in BP alone.
Compared with ARBs, data on the efficacy of ACEi in type 2 DN are less strong, largely because the studies were underpowered or follow-up was short. Nevertheless, some studies did show that ACEi use results in a greater reduction in albuminuria and a slower decrement in renal functional decline compared with other antihypertensive agents. In addition, the Diabetics Exposed to Telmisartan and Enalapril (DETAIL) trial was a randomized controlled trial that compared enalapril to telmisartan in 250 patients with early nephropathy [82]. At 5 years, both groups had similar findings for decline in the GFR, BP, serum creatinine, urinary albumin excretion, ESRD, cardiovascular events, and mortality. The results support the clinical equivalence of ARBs and ACEi in type 2 diabetic patients with nephropathy.
RAASi combinations
On theoretical grounds, a dual blockade of the RAAS with both an ACEi and an ARB should have been superior to monotherapy in treating DN [83]. In the Ongoing Global Endpoint Trial (ONTARGET), combination therapy reduced proteinuria in patients with high cardiovascular risk and prevented new onset of micro- and macroalbuminuria to a greater extent than monotherapy. However, RAASi combination was associated with more end points including the need for acute dialysis, doubling of serum creatinine, and death, than monotherapy [84]. Recently, the Veterans Affairs Nephropathy in Diabetes (VA NEPHRON-D) trial failed to demonstrate a potential for benefit with respect to renal disease progression, mortality, or cardiovascular disease. As in ONTARGET, combined therapy was associated with an increased risk of serious adverse events including acute kidney injury (AKI) and hyperkalemia [85]. The KDIGO guidelines concluded that there is insufficient evidence to recommend combining ACEi with ARBs to prevent progression of CKD [60]. Simultaneous administration of two blockers of the RAAS is currently not recommended in patients with diabetes [86].
The newest RAAS-blocking agent is aliskiren, an oral direct renin inhibitor. In the Aliskiren in the Evaluation of Proteinuria in Diabetes (AVOID) trial, aliskiren patients randomized to aliskiren plus losartan had a significant 20% greater reduction in proteinuria compared to patients randomized to losartan alone, independent of its BP lowering effects [87]. However, the Aliskiren Trial in Type 2 Diabetics Using Cardio-Renal End-points (ALTITUDE) was terminated prematurely because the combination of aliskiren and ACEi or ARB caused increases in nonfatal stroke, hypotension, hyperkalemia, and renal complications [88].
Dosing and adverse effects of ACEi and ARB
The antiproteinuric effect of ACEi and ARBs are at least in part independent of BP reduction, and in individuals, proteinuria may continue to respond to dose escalations beyond those recommended for BP control [89]. Unfortunately, maximal dosing of ACEi or ARBs may be limited by side effects, including hyperkalemia, hypotension, and reduced GFR. Serum creatinine concentration may increase up to 30% in proteinuric patients with renal impairment after an ACEi is started. This rise in creatinine is associated with long-term renoprotection, and therefore the ACEi should not necessarily be stopped in these patients. Increases in serum creatinine concentration above 30% after ACEi initiation should raise the suspicion of renal artery stenosis. Aggressive dose increments of ACEi or ARB, especially in conjunction with diuresis, can precipitate AKI. In advanced CKD, although ACEi and ARBs are not contraindicated, the de novo introduction of these agents or injudicious dose increments may precipitate the need for dialysis prematurely; some caution is appropriate. One small study suggested that in some individuals, RAASi discontinuation late in the course of DN may recover some renal function [90]. The potential for recovering even a small amount of renal function may be especially advantageous when a permanent vascular access is not yet mature, or in cases in which dialysis is inappropriate or unavailable.
Additional interventions
For all diabetic patients, additional therapies beyond glycemic and hypertensive control should be used to reduce the rate of progression of nephropathy and to minimize the risk for cardiovascular events. Indeed, at all stages of CKD, the risk of dying from a cardiovascular complication of diabetes exceeds the risk of progressing to ESRD [91]. Combination therapy includes management of dyslipidemia with a statin, dietary restriction of salt to <5 g/d, lowering of protein intake to ~0.8 g/kg/d in adults with GFR <30 mL/min/1.73 m2, physical activity compatible with cardiovascular health and tolerance (aiming for at least 30 minutes, five times per week), achieving a healthy weight (body mass index 20–25), and smoking cessation.
Novel interventions
Innovative strategies are needed for DN prevention and treatment. Recent trial results have been disappointing. Some trials resulted in an increase in adverse events (aminoguanidine, aliskerin, bardoxolone) [88], [92], [93]. Others may have been abandoned for economic reasons prior to demonstrating benefit (ruboxistaurin; a human monoclonal antibody to connective tissue growth factor) [94], [95]. Some were completed but failed to show benefit (sulodexide) [96], [97]. Others show some benefit in small studies with relatively short follow-up (pirfenidone) [98]. Promising preclinical data suggest that dipeptyl-peptidase-4 antagonists and glucagon-like-1 peptides may attenuate DN independent of their glucose-lowering effects [99], [100]; however, this has not been established in patients [101]. Large-scale clinical trials are needed to confirm safety and to validate the benefits of these agents on relevant clinical end points in DN.
Conclusion
In conclusion, DN is one of the main causes of ESRD and is associated with increased cardiovascular morbidity and mortality. The pathophysiology of diabetes and DN are complex and include interactions between hemodynamic and metabolic pathways, oxidative injury, and cytokines and growth factor elaboration, ultimately leading to renal injury. The current mainstay of pharmacotherapy involves BP control, inhibition of the RAAS with ACEi and/or ARB, and glucose-lowering agents. Disease modifications such as lipid control, dietary restriction, smoking cessation, and weight reduction provide additive renal benefits, particularly in addressing cardiovascular risk. Innovative strategies targeting additional pathophysiological pathways are needed to prevent and treat DN. ClinicalTrials.gov lists more than 500 trials that have been recently completed or are in progress to address DN.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









