



Introduction
Acute kidney injury (AKI) causes significant morbidity and mortality in hospital and outpatient settings [1]. Increased age, comorbidities, and illness severity predispose patients to AKI [2]. While the definition of AKI centers on impaired renal function, AKI also adversely affects the heart, lung, brain, and other organs [3]. Notably, few effective treatments exist for the prevention and treatment of AKI.
Heme oxygenase-1 (HO-1) is an inducible, cytoprotective enzyme that degrades toxic free heme. Heme-containing proteins such as hemoglobin, myoglobin, and cytochromes, among others, contribute to essential physiologic functions [4]. Upon injury, these proteins are destabilized and release free heme, which causes oxidative stress, inflammation, and apoptosis [4]. HO-1 also produces cytoprotective substances including ferritin (co-induced with HO-1 to sequester the released iron), carbon monoxide, and biliverdin (later converted to bilirubin) [5]. HO-2 is the constitutive isoform of HO and is expressed largely in the brain, kidney, and testis.
HO-1 was discovered to be a cytoprotective gene in 1992. This paper reported that HO-1 confers protection in the glycerol model of AKI, in which kidney injury is induced by rhabdomyolysis and hemolysis [6]. This motivated studies in both non-renal contexts and diverse AKI models such as ischemia-reperfusion, cisplatin, and lipopolysaccharide (LPS), which demonstrated the protective role of HO-1 against AKI [7,-11]. While these studies indicated that HO-1 could be a strong candidate for treatment of AKI, further studies are needed to investigate its therapeutic potential. This review focuses on advances in understanding the significance of HO-1 in AKI and efforts to translate HO-1 induction as a therapeutic option for AKI.
Nrf2 activation and AKI
The nuclear factor erythroid 2–related factor 2 (Nrf2) is a key transcription factor involved in regulating HO-1. In healthy individuals, Nrf2 activation is suppressed by Kelch-like ECH-associated protein (Keap1), which promotes Nrf2 degradation. During injury, the Keap-Nrf2 interaction is destabilized, leading to Nrf2 activation. Nrf2 then translocates to the nucleus and upregulates genes, including HO-1. Nrf2 is now identified as a key mechanism mediating protection against AKI, in part through HO-1 induction.
In ischemia-reperfusion injury (IRI) studies, ethyl pyruvate protects against AKI through HO-1 induction via the PI3K/Akt and Nrf2 pathways [12]. HO-1 induction inhibits the production of high mobility group box 1 (HMGB1), a proinflammatory transcriptional regulator. Thus, the protective effects of ethyl pyruvate involve Nrf2 activation and HO-1 induction, culminating in the suppression of HMGB1-dependent inflammation and injury [12].
Studies in cisplatin-induced AKI demonstrate that miR-140-5p, a microRNA (miRNA), is increased and directly targets the 3’UTR of Nrf2 mRNA. This miRNA activates Nrf2 and its target genes, including HO-1. Studies in vitro have demonstrated that miR-140-5p decreases reactive oxygen species (ROS) levels and protects against cisplatin-induced cell death. The protective role of Nrf2 was corroborated by studies involving Nrf2 silencing, leading to loss of the protection conferred by miR-140-5p [13].
Nrf2 also confers protection in hemolysis-induced AKI [14]. Compared to wild-type mice, Nrf2-deficient mice experience more severe AKI and have decreased expression of Nrf2 target genes such as HO-1. Nrf2 activation by sulforaphane ameliorated AKI in both in vivo and in vitro models of heme-mediated cellular injury.
T-lymphocyte-specific activation of Nrf2 protects against AKI [15]. T cells contribute to IRI, but the classic activation mechanism of T cells by alloantigens does not occur in IRI. It is assumed that oxidative stress promotes cytotoxic T cell activation in IRI. Noel et al [15] hypothesized that oxidative stress in IRI activates both cytotoxic T cells and protective T cells. The authors generated mice with amplified Nrf2 in T cells, causing increased baseline antioxidant gene expression, including HO-1 and Nqo1. Nrf2 augmentation in T cells increased intrarenal immune suppressor cells and anti-inflammatory Tregs and decreased pro-inflammatory M1 macrophages. Moreover, mice with T cell Nrf2 augmentation were resistant to IRI.
Preconditioning may also involve Nrf2 activation [16]. Administered before IRI, tin protoporphyrin conferred protection against IRI. Tin protoporphyrin increased intrarenal levels of catalytic iron along with oxidative stress, the latter leading to activation of Nrf2 and its dependent genes.
Oxidant-generating enzymes, such as NADPH oxidase 4 (NOX4), can unexpectedly protect against AKI via Nrf2-dependent mechanisms [17]. NOX4 is expressed in proximal tubules and generates superoxide anion and hydrogen peroxide. Based on studies in NOX4-deficient and WT mice, NOX4 protects against IRI by eliciting Nrf2 activation via its constitutive and baseline oxidant generation.
In addition to protecting against AKI, substantial evidence demonstrates that Nrf2 can mitigate occurrence of the AKI-chronic kidney disease (CKD) transition [18]. Thus, HO-1 induction through the activation of its transcription factor, Nrf2, may protect against AKI in myriad contexts.
HO-1 and preconditioning
Preconditioning involves exposure to an insult that is nephroprotective against subsequent similar or dissimilar insults. In the studies that established the protective effects of HO-1, renal HO-1 induction by hemoglobin protected against AKI and mortality caused by later exposure to heme protein-mediated renal damage [6]. Preconditioning by dissimilar stimuli may also confer protection. For example, endotoxin administered 24 hours before glycerol-induced AKI is protective via HO-1 induction [19].
HO-1 is involved in preconditioning. For example, before IRI, HO-1 induction by a HO-1 fusion protein protects against AKI [20]. Another form of preconditioning involves administration of iron sucrose (an oxidative stress inducer) and tin protoporphyrin (an inhibitor of HO activity). This administration is nephroprotective in ischemic and nephrotoxic models of AKI due mainly to induction of HO-1 and other cytoprotective genes [21]. The same group later compared iron-sucrose/tin protoporphyrin preconditioning with preconditioning by IRI [22].
Timing, dose, and context determine whether prior injury promotes preconditioning or exacerbates injury. For example, administering hemin to mice after bilateral IRI worsens renal damage and oxidative stress [23]. However, hemin administration prior to IRI, in addition to hemin administration post-IRI, protects against AKI.
Nrf2 is upstream of HO-1 and has been investigated as a mechanism for preconditioning. For example, two preconditioning methods to induce Nrf2 activation, IRI and sulforaphane administration, were examined [24]. While both were effective, sulforaphane conditioning exerted greater protection than only IRI and better renal outcomes were achieved when both approaches were employed.
Limb ischemic preconditioning (LIPC) or remote ischemic preconditioning protects against contrast-induced AKI [25]. LIPC treatment prior to contrast administration activated Nrf2, upregulated phospho-Akt, and decreased Nf-κB. Administering a glycogen synthase kinase (GSK)-3β inhibitor recapitulated this protection and may act through activating Nrf2, while suppressing Nf-κB.
HO-1 preconditioning may improve renal transplantation outcomes. For example, administering the HO-1 inducer heme arginate prior to and after renal transplantation upregulates HO-1 in deceased donor transplant recipients [26]. Because heme compounds may lead to adverse as well as protective preconditioning effects, there is a clinical need for strategies to induce HO-1 and reliably confer protective preconditioning. By activating protective mechanisms involving HO-1 induction prior to injury as well as other mechanisms, preconditioning may ameliorate AKI in animal models and may benefit renal transplant recipients and other patients.
HO-1 polymorphisms and AKI
The relationship between the number of guanine and thymine (GT) repeats in the human HO-1 promoter and the incidence of AKI after cardiac surgery was explored by Leaf et al [27]. Prior evidence indicated that relatively long GT repeats, compared with short GT repeats, are associated with less HO-1 inducibility and less HO activity. Leaf et al [27] demonstrated that patients with the long GT repeat polymorphism exhibited higher risk for AKI after cardiac surgery. Increased risk with long GT repeats is also described after kidney transplantation. For example, patients who received kidney transplants from donors with short GT repeats had better outcomes following prolonged cold ischemia after transplantation and after acute rejection, and had lower graft loss caused by chronic allograft nephropathy [28]. Conversely, renal grafts from subjects with long GT repeats exhibited worse renal function two years after transplantation [29].
Bilirubin is a nephroprotective HO product. Lee et al [30] examined the effect of polymorphisms in HO-1 and in the gene responsible for bilirubin degradation, UDP-glucuronosyltransferase (UGT1A1), on renal transplantation outcomes. While the HO-1 A-413T polymorphism did not influence bilirubin levels or transplant outcomes, the UGT1A1*28 polymorphism was associated with higher serum bilirubin levels and improved graft longevity.
In contrast, investigations of pediatric AKI failed to show an association between long GT repeats and AKI risk [31], whereas studies in a Finnish population showed that the short GT repeats were associated with a greater risk for developing sepsis-associated AKI [32].
Other polymorphisms may also be relevant in pediatric AKI: infants with the TT genotype of the 413 T > A polymorphism had a lower occurrence of AKI [31].
Finally, there is no relationship between kidney graft outcomes and specific single nucleotide polymorphisms (rs2071746, rs2071748, rs2071749, and rs5755720) or one microsatellite marker in the HO-1 gene [33]. These studies illustrate that HO-1 gene polymorphisms, such as the long GT repeats in the HO-1 promoter among others, may increase the risk of developing AKI.
HO-1, age, and sex in AKI
Older patients have increased AKI risk and worse AKI outcomes, such as the AKI-CKD transition [34]. Aged mice are also more sensitive to AKI, which may reflect decreased HO-1 expression [35]. Aged mice exhibited worsened renal function and decreased medullary HO-1 induction in response to IRI compared to young mice [35]. When aged mice were administered the HO-1 inducing agent, heme arginate, they demonstrated improved renal recovery after IRI and increased macrophage HO-1 induction. This protection was lost after macrophage depletion using transgenic mice expressing the diphtheria toxin receptor and administered diphtheria toxin. Thus, aged mice may be susceptible to IRI because they lose HO-1 inducibility in macrophages [35]. HO-1 also protects against sensitivity to IRI in young mice, as HO-1 deficiency exaggerates AKI in young mice [36]. The sensitivity of aged HO-1-/- mice to IRI has not yet been explored, but, notably, even without any other insult, aged HO-1-/- mice as compared with aged HO-1+/+ mice demonstrate increased inflammation and cytokine upregulation, suggesting that HO-1 suppresses age-related renal inflammation [36].
Aged mice are also more susceptible to nephrotoxic insults. When young and aged mice were administered hemoglobin, aged mice displayed more severe AKI [37]. While aged mice demonstrated comparable HO-1 induction after hemoglobin, they did not display any HO-2 (the constitutive isoform) mRNA upregulation, in contrast to the moderately increased HO-2 mRNA in hemoglobin-treated young mice. HO-2-/- mice are more sensitive to hemoglobin-induced AKI than HO-2+/+ mice. This study demonstrates the increased susceptibility of aged mice to the nephrotoxicity of hemoglobin, and the role of HO-2 in ameliorating the impact of aging [37].
Females have decreased AKI risk compared to males. A recent study explored the role of HO-2 in the context of IRI, sex, and aging [38]. Aged male HO-2-/- mice displayed worsened renal function after IRI compared to young female HO-2-/-, old female HO-2-/-, and young male HO-2-/- mice subjected to IRI. Phosphorylated STAT3 was markedly induced in the aged HO-2-/- males. pSTAT3 can be a proinflammatory signal and is also upregulated to a greater degree in HO-1-/- compared with HO-1+/+ male mice after IRI [39]. Deficient HO activity, either from HO-1 or HO-2 deficiency, may allow exaggerated pSTAT3 activation after IRI, with attendant inflammation.
Because both age and male sex are AKI risk factors, Boddu et al [40] examined these risk factors by exposing young female, aged female, young male, and aged male mice to cisplatin. Compared to other groups, young female mice were resistant to cisplatin as evidenced by filtration markers, cytokine responses, and autophagy. Interestingly, young female mice exhibited a distinct HO-1 expression profile after cisplatin, which may explain their relative resistance to cisplatin-induced AKI. Consequently, age and sex may affect the risk for AKI, at least partly, through modulating HO-1 expression and HO activity.
HO-1 and myeloid cells in AKI
HO-1 influences inflammation and immunity. For example, HO-1 induces the anti-inflammatory macrophage M2 phenotype, which increases their regenerative ability and IL-10 secretion [41]. HO-1 contributes to dendritic cell (DC) differentiation in the spleen, and promotes the homing of DC precursors [42]. HO activity in antigen-presenting cells promotes Tregs-mediated suppression, which is anti-inflammatory and protective [43].
Prior studies showed that the HO product CO reduces graft immunogenicity after engraftment and reduces chronic allograft nephropathy [44]. Additionally, HO-1 induction or CO administration in organ donors before harvesting decreased expression of MHC class II and co-stimulatory molecules in the recipients’ spleens, suggesting that HO-1 influences DC behavior [45].
HO-1 expression in monocytes/macrophages is beneficial in AKI. Statins induce HO-1 in vascular smooth muscle cells and macrophages. Statin administration reduces IRI-induced AKI, and this protection is blocked by HO inhibition [46]. Statins exaggerated HO-1 induction post ischemia, and infiltrating macrophages were a major source of tissue HO-1 production, leading to the conclusion that statins protect against IRI by inducing HO-1 in circulating monocytes/macrophages in vivo. Ferenbach et al [47] extended these findings in studies involving bone marrow-derived macrophages overexpressing HO-1 using an adenovirus construct. When exposed to LPS and interferon gamma, these macrophages produced increased anti-inflammatory cytokines (IL-10), decreased pro-inflammatory cytokines (TNF-α) and increased phagocytosis of apoptotic cells. HO-1 overexpressing macrophages, administered intravenously, homed specifically to the ischemic kidney and ameliorated AKI.
Myeloid HO-1 expression in IRI was explored by Rossi et al [48]. HO-1 deficiency in myeloid cells in vivo increased histological injury, inflammation, and oxidative stress 24 hours after IRI. Treating WT mice with heme induced HO-1 in the CD11b+F4/80lo myeloid cells (a subset of M2 macrophages), which appeared in the kidney and protected against AKI. Conversely, mice with HO-1 deficiency in myeloid cells were not protected after IRI [48].
Hull et al [49] elucidated how HO-1 regulates myeloid trafficking in AKI using mice with genetic HO-1 alterations, syngeneic transplantation techniques, and visualization of DCs and monocytes in vivo. The authors concluded that HO-1 expression by renal DCs promotes their retention in the kidney where they enhance recovery and decrease fibrosis after IRI. These studies also demonstrate that HO-1 expression in monocytes/macrophages hastens the exit of these cells from the ischemic kidney and their migration to extra-renal sites, thus lessening their involvement in ischemic injury [49].
Using conditional mouse knockouts, Bolisetty et al [50] explored the roles of HO-1 and ferritin using a model of unilateral ureteral obstruction (UUO). HO-1 deficiency increased heavy subunit of ferritin (ferritin heavy chain, FtH) expression and an influx of macrophages with abnormal polarization. Additionally, FtH deficiency in myeloid cells did not affect macrophage migration or polarization, whereas FtH deficiency in the proximal tubules promoted inflammatory cytokines and fibrosis. Thus, FtH in the proximal tubule confers a protective, anti-inflammatory response in the acutely obstructed kidney [50]. Consequently, HO-1 expression by myeloid cells protects against AKI and may influence the behavior and trafficking of these cells during AKI.
Iron dysregulation in AKI
Intracellular catalytic (labile) iron levels are increased in AKI [51,52]. This iron originates from destabilized intracellular heme proteins such as cytochrome p450 and from injured mitochondria. Catalytic iron causes injury via the Haber-Weiss reaction, which generates toxic hydroxyl radicals. In human AKI, plasma labile iron levels are upregulated and are implicated in AKI and its extrarenal effects [51,52]. Catalytic iron contributes to ferroptosis, a form of cell death different from apoptosis and necrosis. Ferroptosis is regulated by ROS and lipid peroxidation and is characterized ultrastructurally by the compaction of mitochondria and their membranes.
HO-1 activity releases iron, which raises the question of whether HO-1 drives or protects against ferroptosis. Adedoyin et al [53] addressed this question using proximal tubular cells (PTCs) from HO-1+/+ and HO-1-/- mice. Exposing renal PTCs to ferroptosis inducers (erastin and RSL3) increased cell death and HO-1 induction. Such cell death increased in PTCs with genetic HO-1 deficiency, and worsened when these HO-1 deficient cells were exposed to iron. The death of HO-1+/+ and HO-1-/- cells induced by erastin was attenuated by iron chelation, antioxidants, and a ferroptosis inhibitor. Thus HO-1 deficiency promotes ferroptosis, and HO-1 protects against ferroptosis through its antioxidant products.
Ferroptosis inhibition by HO-1 was demonstrated in AKI studies examining Pannexin-1 (PANX-1), an ATP-releasing pathway family protein that initiates apoptosis in other tissues [54]. In these studies, PANX-1 deletion protects against ischemic AKI by reducing ferroptotic cell death. In vitro, PANX-1 deletion decreased ferroptosis, lipid oxidation, and iron aggregation caused by erastin. The protective effects of PANX-1 deletion involved HO-1 upregulation, which decreased ferroptinophagy through MAP kinase system signaling.
Zarjou et al [55] illustrated the importance of iron-binding and iron-transporting proteins in AKI. In glycerol and cisplatin induced AKI, FtH deletion in renal proximal tubules (PTs) exacerbated renal injury, increased mortality, and heightened apoptosis. FtH comprises the iron-binding protein, ferritin, and exhibits ferroxidase activity that blocks iron redox cycling. The heightened injury in both AKI models occurred despite HO-1 upregulation [55]. Furthermore, expression of the iron-transporting protein, ferroportin, was suppressed in mice with proximal tubule specific FtH deficiency, whereas FtH chain induced ferroportin. Thus, the cytoprotective effects of FtH involve the binding of catalytic iron in ferritin, interrupting the redox cycling of iron and accompanying oxidative stress, and exporting labile iron out of the kidney via ferroportin.
FtH also protects against acute glomerular injury during HO-1 and FtH coexpression [56]. In companion in vitro studies, HO-1 inhibition decreased FtH expression and worsened mesangial cell death in cultured mesangial cells. HO-1 induction conferred resistance to hydrogen peroxide, and FtH knockdown inhibited this protection. Conversely, RNA interference of HO-1 increased sensitivity to oxidant injury, which was overcome by FtH upregulation.
Iron dysregulation in AKI also involves hepcidin [57]. Hepcidin prevents cellular iron export by causing ferroportin degradation. IRI causes ferroportin induction in the liver and spleen, leading to iron export from these organs to systemic circulation and kidney. Hepcidin administration decreases this iron transport by causing ferroportin breakdown. Hepcidin thus protects against IRI by reducing iron delivery from the liver and spleen to the kidney and by increasing renal ferritin content. Additionally, hepcidin protects against AKI by increasing renal ferritin content, which Zarjou et al [55] demonstrate is a nephroprotectant. That hepcidin-/- mice exhibited increased sensitivity to IRI, which is lessened by hepcidin protein administration, is evidence of the nephroprotective effects of hepcidin [57]. These studies indicate that HO-1 may counteract the damaging effects of iron dysregulation in AKI through decreasing ferroptosis and promoting the binding and safe transport of free iron through its relationship with FtH, among other effects.
HO-1 and sickle cell disease
Sickle cell disease (SCD) is caused by two mutant hemoglobin genes with at least one being the sickle β-globin gene [58]. A hallmark of SCD is vaso-occlusion caused by the sickling of red blood cells (RBCs). Vaso-occlusion is accompanied by sickle crisis episodes, and these episodes lead to hemolysis, chronic ischemia, and organ injury. Hemolysis and systemic inflammation are notable characteristics of SCD. SCD can also provoke both AKI and CKD.
In 2001, Nath et al [59] discovered the crucial link between SCD, oxidative stress, and HO-1 induction. In a transgenic mouse model of SCD, intrarenal lipid peroxidation and heme were increased, and HO-1 was induced in the kidney. Extrarenal tissues such as the pulmonary vasculature and circulating endothelial cells also expressed increased HO-1. Oxidative stress was implicated as a cause of RBC sickling because, when oxidative stress was exacerbated, RBC sickling and vaso-occlusion were significantly worsened in the kidney.
Several studies have demonstrated that HO-1 protects against vaso-occlusion in extrarenal vascular beds. For example, induction of Nrf2 and HO-1 ameliorates vaso-occlusion in the dorsal skin fold circulation [60]. In other studies, this vaso-occlusion is decreased by either haptoglobin (which binds free plasma hemoglobin) or hemopexin (which binds free plasma heme), with both proteins conferring protection via HO-1 upregulation [61]. HO-1 expression by patrolling monocytes reduces vaso-occlusive disease in SCD [62]. In SCD, intravascular hemolysis produces free hemoglobin and heme, which damage the endothelium. In addition to removing damaged endothelium, patrolling monocytes induce HO-1, which exerts anti-inflammatory and anti-adhesive effects and attenuates vaso-occlusive disease.
In SCD, the kidney is more sensitive to IRI [59,63]. Unexpectedly, pretreatment of sickle mice with a HO activity inhibitor reduces this increased IRI sensitivity [64]. There are two potential explanations for this finding. First, HO-1 may cause injury in this context because increased iron release by HO activity in the heme-enriched kidney in SCD may foster oxidative stress during IRI. Increased iron release may overwhelm mechanisms for binding or transporting iron and, thus, inhibition of HO activity may be protective. Alternatively, competitive HO inhibitors are known to increase HO-1 protein expression [65]. Regardless of HO activity, increased HO-1 protein can be protective through signaling pathways [66]. Additionally, chronic administration of a HO inhibitor reduced renal tubulointerstitial disease and vaso-occlusive disease in murine SCD. Similar to the findings from the acute IRI experiments, these chronic studies suggest that oxidative stress is increased in the kidney in SCD, and inhibition of iron release from renal heme may reduce iron involvement in oxidative stress. In other studies, HO-1 and ferritin H chain are chronically upregulated by weekly administration of haptoglobin, but the functional significance of such upregulation is uncertain [67].
Saraf et al [68,69] evaluated the relationship between HO-1 gene polymorphisms involving long versus short GT repeats in SCD and AKI. Along with a lower estimated glomerular filtration rate, patients with long GT repeats are more susceptible to AKI and the presence of hemoglobinuria. Thus, HO-1 and HO activity are highly induced in the kidney in addition to other tissues in SCD. While this induction exerts cytoprotective effects in other tissues, the functional significance of this induction in the kidney necessitates further study beyond a HO chemical inhibitor approach.
HO-1 and malaria
Malaria, a disease caused by Plasmodium species, constitutes a major cause of morbidity and mortality in underdeveloped countries. In the first phase of infection, Plasmodium species infect the liver, while the second phase involves infection of RBCs. This later phase displays systemic manifestations and end organ damage such as AKI and cerebral malaria.
Seminal studies demonstrated that HO-1 is induced in wild type mice infected with Plasmodium berghei ANKA [70]. Mice deficient in HO-1 or subjected to HO inhibition developed an increased incidence of experimental cerebral malaria (ECM). Conversely, HO-1 induction or CO administration reduced ECM incidence. Neither strategy lowered parasitemia, but each one decreased blood-brain barrier (BBB) disruption, neurovascular RBC sludging, and inflammation. Based on such studies [70,71], the authors conclude that high free plasma heme levels, caused by the oxidation of plasma hemoglobin released by lysed infected RBCs, causes BBB disruption. While HO-1 counteracts plasma heme elevations, CO binds hemoglobin and prevents its oxidation to produce free heme.
Subsequent studies showed that HO-1 protects against noncerebral manifestations of malaria [72], and that HO-1 upregulation in organs mitigates increased plasma heme [73]. This group focused on the kidney and its HO-1 induction in response to hemoglobin and heme. Both HO-1 and ferritin H chain upregulation occurs via the Nrf2 pathway in experimental malaria, which degrades heme and reduces AKI risk. Thus, this inductive pathway confers tolerance and reduced renal injury without decreasing parasitemia.
Other studies explored the relationship between STAT3, CXCL10/CXCR3, and HO-1 in cerebral malaria [74]. CXCR3 receptor activation by its ligand CXCL10 heightens apoptosis, chemotaxis, angiogenesis, and cell growth and contributes to severe malaria in animal models. Heme induces both HO-1 and CXCL10 through STAT3 activation in the P. berghei ANKA model of cerebral malaria and in vitro studies. Furthermore, HO-1 and CXCL10 may share a regulatory feedback loop in which CXCL10 activates HO-1 while HO-1 downregulates CXCL10, thereby suppressing a harmful pathway in malaria.
Finally, chronic HO-1 induction in malaria may not be universally protective. Specifically, chronic HO-1 induction may predispose to bacterial infections because HO-1 inhibits neutrophil infiltration and responses. Mooney et al [75] revealed persistent hemolysis and increased HO-1 among children with asymptomatic malaria, along with higher IL-10 levels, which upregulates HO-1. Malaria is associated with increased bacterial infections, including Salmonella infections. The suppressive effects of HO-1 on neutrophil behavior may predispose to susceptibility to bacterial infection. Together, these studies provide strong evidence that the acute induction of HO-1 during malaria reduces the accumulation of plasma heme released by lysed RBCs and accompanying inflammation and thus ameliorates the renal damage associated with this disease.
Sepsis, HO-1, and AKI
Studies employing HO-1-/- mice demonstrate that these mice, compared with HO-1+/+ mice, when subjected to LPS-induced sepsis, exhibit exaggerated NF-κB activation, upregulation of both Th1 and Th2 cytokines and chemokines, lymphocyte apoptosis, and renal functional impairment [9]. These studies established the role of HO-1 as a protectant against sepsis-induced AKI.
Recent studies have sought agents that may protect against sepsis-induced AKI by inducing HO-1. Kang et al [76] assessed renal thrombotic microangiopathy (TMA) as an indicator of sepsis severity. They reasoned that HO-1 induction may protect against TMA because of the known anticoagulant effects of HO-1 partially due to inducing activated protein c (APC). As a potent anticoagulant, APC is generated from protein c (PC) by thrombomodulin; when PC is bound to the endothelial cell protein c receptor (EPCR), such APC generation is accelerated. In the cecal ligation and puncture (CLP) model of sepsis, TMA was accompanied by diminished renal TM and EPCR expression. Kang et al [76] demonstrated that HO-1 induction by hemin diminished TMA while increasing renal expression of the anticoagulant components, TM, and EPCR. Additionally, inhibiting HO activity decreased TM and EPCR expression. Inducing HO-1 may thus reduce sepsis-induced TMA by promoting APC induction, an anticoagulant and anti-inflammatory species.
Resveratrol induces HO-1 in the vasculature. In the CLP model, resveratrol improves renal function and downregulates inflammation [77]. In an in vitro model of sepsis based on LPS-treated renal epithelial cells, resveratrol activated Nrf2 and its target genes, including HO-1, while decreasing LPS-induced inflammation. Nrf2 knockdown by siRNA in vitro attenuated the suppressive effects of resveratrol on inflammation. Thus the beneficial effects of resveratrol in sepsis may involve the Nrf2/HO-1 pathway.
Novel approaches that protect in sepsis models by reducing the inflammatory response and improving renal function also induce HO-1. For example, in the CLP model, such findings have been reported for neutrophil gelatinase-associated lipocalin [78], pachymic acid [79], and honokiol (components of traditional Chinese medicines) [80]. Similarly, in the LPS model, 3,4,5-trihydroxycinnamic acid, a tea extract, is protective and induces HO-1 [81]. However, the contribution of HO-1 thus induced in these studies has not been specifically demonstrated.
The anti-inflammatory effects of HO-1 can be harnessed by cell therapy. For example, in the CLP model, administration of mesenchymal stem cells that overexpress HO-1 decreases renal injury and mortality [82]. Thus, sepsis induces HO-1, and this induction is generally a protective response in the kidney and other organs.
HO-1 and autophagy
Autophagy removes injured organelles by engulfing and degrading them through the involvement of lysosomal enzymes [83]. This process may be organelle-specific as, for example, mitophagy degrades injured mitochondria. During health, autophagy occurs in a low-grade manner in the healthy kidney because it is part of the homeostatic process that preserves renal integrity during aging. In AKI, autophagy is generally nephroprotective since AKI is worsened in experimental models when autophagy is impaired or abrogated. A complex relationship exists between autophagy and apoptosis, with certain proteins participating in specific segments of the autophagic and apoptotic pathways [83].
Using both in vivo and in vitro approaches as well as strategies that genetically delete or overexpress HO-1, Bolisetty et al [84] analyzed in-depth the significance of HO-1 in cisplatin-induced AKI. In proximal tubular epithelial cells lacking HO-1, oxidative stress is increased even without an imposed insult, leading to organelle damage. Basal autophagy is increased, reflecting a homeostatic mechanism that removes injured organelles due to increased basal oxidant stress. When cisplatin is administered with the HO-1 system intact, oxidant stress is imposed largely by increased free heme present in cisplatin-treated kidneys; oxidant stress is heme-driven and autophagy also occurs. When cisplatin is administered in HO-1 deficiency, autophagy is increased but is dysregulated and has impaired progression with more abundant apoptosis. These changes are attenuated when cells regain their HO-1 expression. Bolisetty et al [84] conclude that HO-1 is protective because it decreases elevations in free heme and oxidant stress and the autophagic response, enabling autophagy to be a regulated process and activated as needed.
Cyclosporine can cause human AKI. When renal epithelial cells are exposed to cyclosporine, they exhibit oxidative stress, apoptosis, and autophagy [85]. The compound schisandrin B protects against cyclosporine-induced cell death by inducing Nrf2 and HO-1 and by mitigating oxidative stress, apoptosis, and autophagy [85].
The interaction of Toll-like receptors (TLR2 and TLR4), cisplatin-induced AKI, and HO-1 has been recently studied [86]. In response to cisplatin, TLR2 deficiency worsens renal injury, while these effects were not observed in TLR4-deficient mice. Furthermore, the TLR2-deficient mice exhibited a less developed autophagic response, and hemin, administered to TLR2- and TLR4-deficient mice, ameliorated renal injury, but less so in TLR2 KO mice. The authors conclude that TLR2 protects against cisplatin-induced AKI, possibly through its induction of autophagy and its relationship with HO-1 [86].
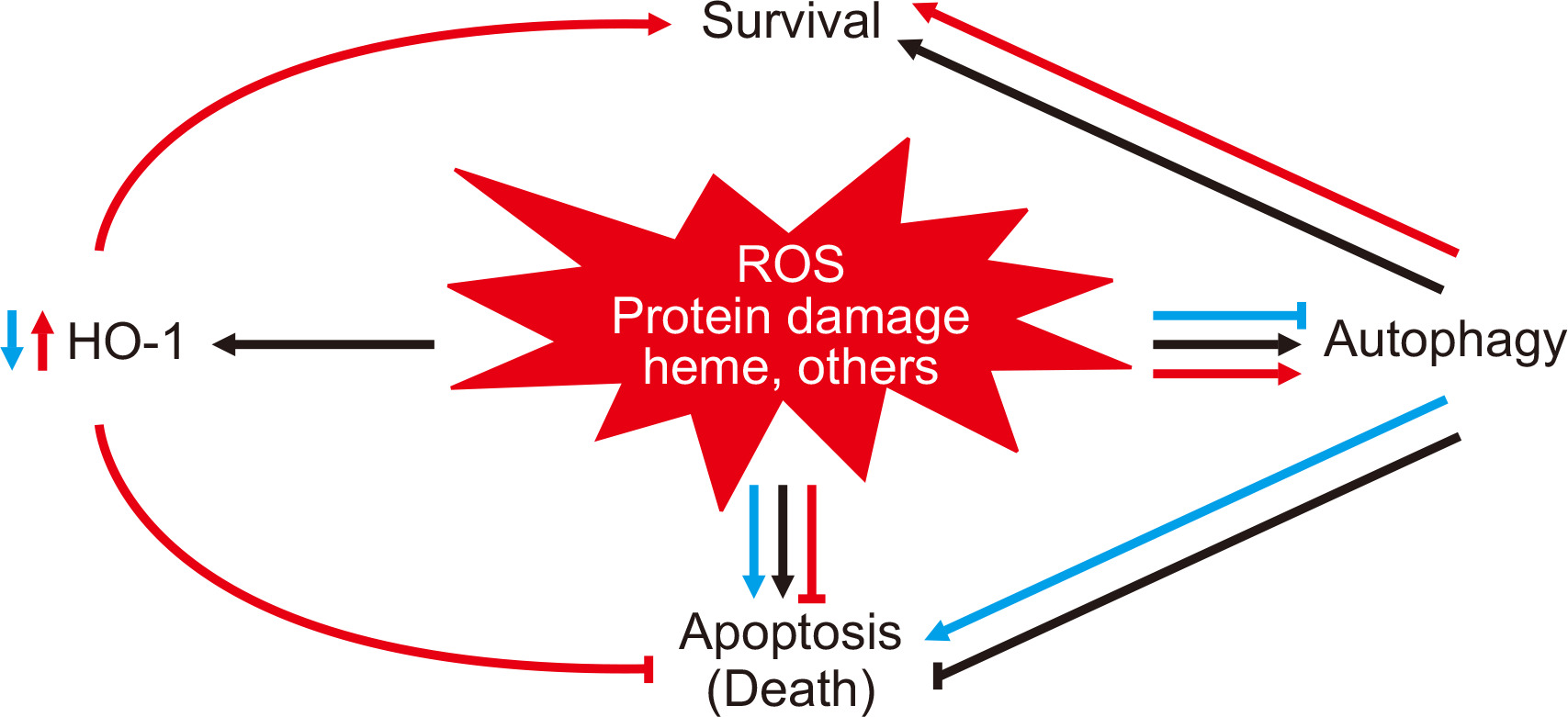
The relationship between HO-1 and autophagy during cisplatin injury and the consequences of either HO-1 deficiency or overexpression in this setting are shown in Fig. 1 [84]. Consequently, HO-1 induction leads to the reduction of oxidative stress and free heme, which may promote the regulation of autophagy and thus mitigate potentially harmful effects during AKI.
HO-1 and the AKI to CKD transition
Studies conducted 20 years ago considered whether AKI and CKD are truly separate renal syndromes [87]. The authors questioned if mechanisms identified in the development of AKI could cause CKD in the right context [87]. In these studies, rats were repetitively exposed to heme proteins and demonstrated a triphasic response: AKI after the first dose, resistance to subsequent exposure, and, finally, CKD driven by chronic monocyte chemoattractant protein (MCP)-1 and transforming growth factor (TGF)-β upregulation. Because heme proteins upregulate HO-1, these authors examined the role of HO-1 in this AKI-CKD transition by subjecting HO-1+/+ and HO-1-/- mice to repeated hemoglobin exposure [88]. HO-1-/- mice exhibited exaggerated chronic inflammation, caused largely by NF-κB activation and its upregulation of downstream MCP-1.
The role of HO-1 in the AKI-CKD transition and in the epithelial to mesenchymal transition (EMT) was explored in the context of chronic inflammation caused by acute obstruction of the urinary tract. While the field of EMT is controversial, EMT clearly occurs in vitro and at least “partial” EMT occurs in vivo in that epithelial cells lose their phenotype and behave like mesenchymal cells. Kie et al [89] demonstrated that features of EMT and tubulointerstitial disease are exaggerated in HO-1-/- compared to HO-1+/+ mice in the UUO model. These findings indicate that HO-1 suppresses EMT and the contribution of EMT to the AKI-CKD transition. This is supported by in vitro studies; for example, Clark et al [90] demonstrated that simvastatin inhibited EMT of renal proximal tubule cells through a HO-1-dependent pathway that decreased oxidant stress.
Maladaptive repair after repeated acute injuries can lead to CKD development, and Nrf2 activation (and downstream HO-1) may lessen this maladaptive repair [91]. In the folic acid model of the AKI-CKD transition, Lu et al [92] observed decreased nuclear Nrf2 and downstream genes such as HO-1. In vivo, inhibiting GSK3-β reduced the AKI-CKD transition and restored the cytoprotective effects of Nrf2, suggesting that GSK3-β, independent of Keap-1, downregulates nuclear Nrf2 and its protective effects in this model. Similarly, the tea extract epigallocatechin-3 gallate, a potent antioxidant, can retard the AKI-CKD transition, at least partially through the Nrf2-HO-1 pathway [93].
Epigenetic modifications to proinflammatory genes may influence the shift towards chronic inflammation in the AKI-CKD transition. Zager and Johnson [94] examined histone modifications on proinflammatory TNF-α, MCP-1, and TGF-β1 genes, which occurred right after the induction of the glycerol model of AKI, and then steadily increased, even though resolution of AKI began. While HO-1 upregulation occurs in the model, regression in such expression antedated the proinflammatory response, thereby shifting to a proinflammatory profile. The authors speculated that these prompt and progressive histone modifications, unopposed by now normalizing HO-1 expression, underlie heightened inflammation after AKI and the risk of AKI-CKD transition. Thus, HO-1 expression may decrease the risk of developing CKD following AKI in multiple ways, including by inhibiting EMT, activating the transcription factor Nrf2, and preventing histone modifications that promote proinflammatory gene expression.
Conclusion
As summarized in the present review and also in a table from our prior review [1], there is incontrovertible evidence for the beneficial effects of HO-1 and its products in AKI patients (Table 1) [1,10,36,47,49,50,84,95,-100]. HO-1 protects against diverse insults imposed on multiple organs, including the kidney. As highlighted in this review, HO-1 induction and/or delivery of its products can protect against AKI whether AKI is caused by ischemic or nephrotoxic insults or sepsis. In human AKI as well as preclinical AKI models, HO-1 is robustly induced; strategies that either negate or upregulate HO-1 expression either worsen or protect against AKI, respectively. However, several obstacles remain in translating these observations to patient care. For example, AKI often occurs with little warning and most evidence showing the protective effects of HO-1 involves its induction or administration of its products before AKI occurs. Evidence is needed that HO-1 upregulation or administration of its products after the insult are also beneficial. Another challenge is that HO-1 is commonly induced by stimuli that produce injury. The development of non-toxic HO-1 inducers such as those that upregulate HO-1 by Nrf2 activation could provide critical progress in this area. Finally, significantly increased levels of HO-1 products such as bile pigments and CO can be toxic. The threshold at which HO-1 upregulation transitions from providing beneficial to toxic effects in the dose response relationship is yet to be elucidated. Despite these obstacles, investigating the treatment potential of HO-1 remains a promising opportunity to counteract the damaging consequences of AKI and thereby improve outcomes in patients with AKI.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









