


Introduction
Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are one of the most common and important causes of nephrotic syndrome (NS), accounting for >75% of NS cases in children [1]. In adults, MCD and membranous nephropathy (MN) are most frequent causes of idiopathic NS [2]. Heavy proteinuria, hypoalbuminemia, edema, and related complications such as thromboembolism, infection, and malnutrition are typical symptoms and signs of NS; in particular, a large amount of protein in the urine often leads to progression to end-stage renal disease. In addition, proteinuria is an indicator of treatment response and important long-term prognostic marker in renal disease progression. Increasing evidence suggests that podocyte injury plays an important role in the development of proteinuria and pathogenesis of various glomerular diseases, including MCD, FSGS, and MN. Podocytes are terminally differentiated and cover the urinary surface of glomerular basement membrane. Foot process from one podocyte forms interdigitation with the foot processes of adjacent podocytes to form the slit diaphragm (SD), which functions as the ultimate filter with 4- × 14-nm-sized pores. Since the discovery of mutation of NPHS1 gene, which codes for nephrin (an SD-associated protein), in patients with Finnish-type congenital NS, mutations of several podocyte-associated genes including CD2AP, TRPC6 [17], NPHS2, and NEPH1 were found to be associated with NS [3,4]. Podocytes and SD-associated molecules have therefore become an important target for therapeutic interventions in proteinuric kidney diseases. Synaptopodin is an SD-associated protein, which maintains podocyte integrity. Dephosphorylation or ubiquitination (or in some cases both) of synaptopodin induces derangement of actin cytoskeleton, which results in foot process effacement [5]. Immunologic and metabolic stimuli including activation of cytokine- and calcineurin-dependent mechanisms lead to degradation of synaptopodin and podocyte injury [6].
Various immunosuppressive agents have been widely used to treat glomerular diseases and the effects of these drugs were thought to be solely immune mediated [7,8]. However, during the past decade, advances in podocyte biology and pathogenesis of proteinuric disease unveiled new molecular players responsible for the development of proteinuria; in addition, unexpected mechanisms of action of widely used immunosuppressive agents that are independent of their traditional immunomodulatory function have been discovered [9].
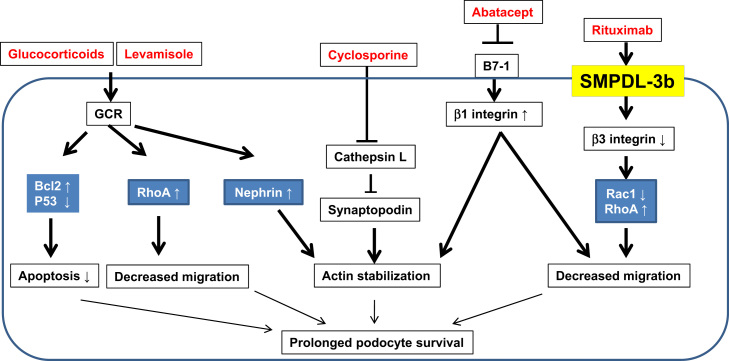
In this mini review, we describe the main targets of immunosuppressive agents in podocytes and review their mechanisms of action independent of immunological function. Furthermore, we also suggest potential new targets for drug development in podocytes. Because side effects develop in a high proportion of patients with prolonged and high-dose immunosuppressive treatment, it is important to understand the optimal doses and target of immunosuppressive agents, as low doses or specific targeted therapy may be more beneficial in patients with proteinuric kidney diseases. Fig. 1 shows a schematic diagram for nonimmunologic targets of immunosuppressive agents in podocytes. Potential targets of immunosuppressive agents in podocytes are given in Table 1.
Evidence of immunologic mechanisms in idiopathic NS
Shalhoub [10] hypothesized that MCD is associated with lymphocyte-derived permeability factor and increasing evidence suggests that cell-mediated immune systems are activated in MCD. Development of MCD is coincidental in Hodgkin’s lymphoma, which is a T-cell disorder [11]. Incidence of atopy, which is also associated with cell-mediated hypersensitivity, is higher in patients with MCD [12]. Idiopathic NS responds to immunosuppressive agents such as corticosteroid and cyclosporine to suppress cell-mediated immunity. In addition, immunologic mechanisms are supported based on several clinical observations that relapse of MCD is experienced after viral or bacterial infections, inhaling allergens, and vaccination. Some studies reported that T cells are clonally expanded and cytokines derived from activated T and B cells are elevated in patients with idiopathic NS [13,14]. van den Berg and Weening [15] also demonstrated that interleukin (IL)-10 and IL-13 as cytokines produced by T lymphocytes are elevated in patients with MCD. IL-10 and IL-13 were normalized after remission of NS, whereas the protein concentrations and messenger RNA (mRNA) levels were upregulated again following NS relapse [15]. In addition, autoantibodies to PLA2R1 bind to epitopes on specific domains of PLA2R1 expressed on the podocyte surface and this phenomenon is demonstrated as a key pathogenetic mechanism in idiopathic MN [16,67]. However, cyclosporine therapy reduced proteinuria in human and experimental Alport’s syndrome models and in patients with NPHS2 mutation and nonimmunological and genetic glomerular diseases. Although cyclosporine decreased proteinuria in patients with MN, repeat kidney biopsy results showed many large electron-dense immune deposits [18–21]. Recent studies also demonstrated that circulating permeability factors are related to the development of NS [22]. These observations suggest that the action of these agents might be beyond immune mechanisms.
Nonimmunologic targets of immunosuppressive agents in podocytes
Glucocorticoids
Glucocorticoids has been widely used for many years and is the standard first-line drug for patients with MCD and FSGS; however, their mechanism of action or target cells in the kidney in this group of patients remains unclear. Glucocorticoid suppresses cell-mediated immunity by blocking the action of cytokines including IL-2, and subsequently reducing T-cell proliferation. These effects of glucocorticoids also diminish humoral immunity by suppressing B-cell clonal expansion and antibody production. However, puromycin aminonucleoside (PAN)-induced NS, which is a well-described model of MCD and FSGS, has no evidence of immunologic mechanisms, and glucocorticoids ameliorates proteinuria in PAN-induced nephrosis. In addition, glucocorticoids exerts its action by binding to the intracellular glucocorticoids receptors (GCRs), which are present in glomerular cells including podocytes. Glucocorticoids attenuates podocyte apoptosis in PAN-induced podocyte injury by restoration of Bcl-2 and reduction of p53 in PAN-treated podocytes [23,24]. Glucocorticoids also prevents PAN-induced translocation of apoptosis-inducing factor. Another study showed that glucocorticoids upregulated nephrin and downregulated vascular endothelial factor in podocytes. In addition, glucocorticoids downregulated cyclin kinase inhibitor p21 and IL-6 expression and augmented podocyte survival [25]. Glucocorticoids in podocytes not only reduces apoptosis, but also enhances recovery by stabilizing actin filaments and maintaining podocyte survival. This protective effect against actin-cytoskeleton depolymerization in podocytes is glucocorticoids specific and could be mediated by increase in the activity of Rho-A (a small guanosine triphosphatase protein), which plays an important role in stress fiber stabilization [26]. More recently, the possibility that ACTH may directly target podocytes via melanocortin receptor 1 has also been studied [66].
Levamisole
Levamisole was a synthetic imidazole derivative used for its anthelmintic effects. Because adverse effects including agranulocytosis have been reported, levamisole is no longer used to treat parasitic infections. However, this drug has an immune-modulating action and has been used in the treatment of some cancers such as colon cancer and melanoma [27,28]. Levamisole, which modulates type 1 and type 2 immune responses through enhancing IL-18 activity, has also been used for the treatment of NS in children [8,29]. A recent study [30] also reported that levamisole is able to induce expression of GCR activities and to activate GCR signaling in human podocytes, suggesting that levamisole can also directly affect the targets in podocytes.
Calcineurin inhibitors
Calcineurin inhibitors including cyclosporine A (CsA) are widely used in patients who received organ transplantation. It is also effective for patients with various glomerulopathies including refractory NS, for both the steroid-dependent relapsing (steroid-dependent nephrotic syndrome) and the steroid-resistant (steroid-resistant nephrotic syndrome) types [31]. CsA is an option for patients who have not responded to conventional steroid treatment and also may now serve as a first-line immunosuppressant for treating children with refractory NS. Cyclosporine inhibits nuclear factor of activated T-cell signaling, which results in inhibition of calcineurin activity in T-cells [32]. CsA also reduced cytokine production and effectively suppressed the T-cell function. The mode of action of cyclosporine in reducing proteinuria is not merely linked to its immunosuppressive effects, as a reduction of proteinuria following cyclosporine therapy is also observed in nonimmunologic glomerular injury such as Alport’s syndrome and PAN-induced nephrosis. In addition, low doses of CsA, which showed ineffective immunosuppressive action, also reduced proteinuria in MCD and MN. A previous report by Faul et al [33] revealed that podocyte is a direct target of CsA. Calcineurin-mediated dephosphorylation of synaptopodin leads to degradation by cytosolic cathepsin L, which subsequently results in destabilization of podocyte actin cytoskeleton and changes in motile phenotype of podocytes. CsA induces synaptopodin to bind to the 14-3-3 site and stabilizes (phosphorylates) synaptopodin. Consequently, the phosphorylated synaptopodin becomes refractory to degradation by cathepsin L. In this mechanism, CsA stabilizes the stress fiber of podocyte and prolongs podocyte survival. This suggests that even lower doses of CsA would be effective in reducing proteinuria. Transient receptor potential channel 6 (TRPC6) in podocytes enhances calcium influx and increases their motility. TRPC6 overexpression has been observed in patients with familial-type FSGS as well as acquired forms of NS [17,34,35]. CsA was reported to downregulate TRPC6 expression in an animal model of FSGS [36]. CsA can also induce vasoconstriction in the afferent arterioles in the kidneys, causing changes in surface charge and permselectivity of the glomerular filtration barrier [37].
Rituximab
Rituximab is a chimeric monoclonal antibody that binds to the B-cell surface antigen CD20 and depletes B-cell lineage [38]. Rituximab also binds to acid sphingomyelinase-like phosphodiesterase 3b (SMPDL-3b) protein, which induces acid sphingomyelinase activity and mediates growth inhibition on B cells [39]. This drug has been used for the treatment of B-cell-proliferating hematologic malignancies and antibody-associated acute rejection in kidney transplantation [38]. Because complete remission of NS unexpectedly occurs in patients with post-transplant recurrent FSGS treated with rituximab, this monoclonal antibody has been a promising candidate drug for treating recurrent or refractory FSGS [40]. Rituximab was occasionally effective to treat some cases of post-transplant recurrent FSGS and refractory FSGS in uncontrolled studies. However, little is known about B-cell-associated mechanisms in FSGS [41,42]. A recent study revealed that human glomeruli including podocytes expressed neither CD20 mRNA nor protein, and rituximab restored actin-cytoskeletal derangement through B-cell-independent mechanisms. A previous study demonstrated decreased glomerular SMPDL-3b expression in patients with recurrent FSGS after kidney transplantation and reduced acid sphingomyelinase activity in podocytes treated with sera from these patients [43]. A recent study also demonstrated that glomerular SMPDL-3b mRNA expression was decreased in FSGS patients. In podocytes treated with sera of high-risk FSGS patients, podocyte apoptosis and loss of stress fiber were observed [44]. Sera of patients with recurrent FSGS after kidney transplantation induce actin-cytoskeleton derangement. The restoration of SMPDL-3b expression in podocytes by rituximab prevented the disruption of stress fiber and podocyte apoptosis. An ongoing randomized clinical trial conducted at the University of Miami, Miami, FL, USA, will help determine whether rituximab can prevent FSGS recurrence after kidney transplant and to determine whether such protection is associated with a modulation of SMPDL-3b expression in podocytes.
Abatacept B7-1-blocking agents
B7-1 (CD80) and B7-2 (CD86), known as costimulatory molecules, are transmembrane proteins usually expressed on the surface of antigen-presenting cells (macrophages and B cells) and bound to CD28 constitutively expressed on T cells [45]. CD80 and CD86 provide costimulatory signals to regulate T-cell-mediated immunity by the production of various ILs, especially IL-2 and IL-6. Costimulatory modulators have been developed and highlighted as promising agents for maintaining immunosuppression in organ transplantation and for treatment of rheumatoid arthritis [46,47]. Meanwhile, B7-1 and B7-2 are also expressed in glomeruli. In particular, B7-1 is abundantly expressed in podocyte and its expression is enhanced in experimental primary glomerular disease. In addition, urinary B7-1 concentrations in patients with MCD were increased. B7-1 in urine returned to the normal range during remission but re-elevated during the relapse in these patients. Reiser et al [48] also clearly demonstrated the role of B7-1 in derangements of actin cytoskeleton in podocytes. They showed that lipopolysaccharide (LPS), which is an endogenous inducer of B7-1 through Toll-like receptor-4 signaling, upregulated actin-cytoskeletal reorganization and induced foot process effacement and proteinuria, but knocking-down of B7-1 ameliorated LPS-induced cytoskeletal changes in podocytes. These changes are also observed in severe combined immunodeficiency mice, which suggested that the action of B7-1 in podocyte injury is associated with T- and B-cell-independent mechanisms. Galiximab, an anti-CD80 monoclonal antibody, has been used for treatment of hematologic malignancy such as relapsed non-Hodgkin’s lymphoma [49,50]. Recently, Yu and co-workers [51] demonstrated that B7-1-blocking agents (abatacept) may become considerable therapeutic agents in patients with FSGS. B7-1-positive podocytes showed near-complete loss of β1 integrin activation and increase in motility. However, abatacept restored β1 integrin activation and reversed podocyte motility. In addition, abatacept treatment was associated with partial or complete remission of proteinuria in a very small series of patients with FSGS or recurrent FSGS after renal transplantation. Should these data be confirmed in prospective randomized clinical trials, it is possible to envision the development of personalized therapeutic decisions. In particular, patients with proteinuria would be classified as either B7-1-positive or B7-1-negative kidney disease in their renal biopsy samples. B7-1-blocking agents might be potential therapeutic agents in patients with FSGS, and therefore, additional clinical studies are needed in this regard.
Sphingosine-1-phosphate agonist: FTY720
FTY720, which is an immunosuppressive B7-1 positive agent approved for treatment of multiple sclerosis, might be a potential candidate for the treatment of podocyte injury. Sphingosine-1-phosphate (S1P) is a biologically active sphingomyelin metabolite and is phosphorylated by sphingosine kinase-1. S1P exerts its action by binding to five structurally related G-protein-coupled receptors (S1P1–S1P5 receptors) [52]. Because S1P sequesters lymphocytes and inhibits T-cell chemotaxis and T-cell proliferation [53], the S1P agonist FTY720 is proposed to treat transplant rejection and autoimmune disease such as multiple sclerosis [54,55]. Besides, FTY720 has a protective effect on ischemia-reperfusion animal models and improved diabetic kidney injury without alterations of T and B cells [56,57]. In addition, a selective S1P agonist, SEW2871, abolished high glucose (HG)-induced vascular endothelial growth factor and tumor necrosis factor-α upregulation in podocytes, and also improved proteinuria through restoration of podocyte-specific molecules such as nephrin and podocin in streptozotocin-induced diabetic nephropathy. These data indicate that the S1P agonist directly affected podocytes and also might be a therapeutic candidate in diverse podocytopathic diseases.
Mammalian target of rapamycin inhibitors
Rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, inhibits mTOR activity, eventually leading to cell cycle arrest at the G1 phase [58]. mTOR inhibitors impair T-cell proliferation and are used for immunosuppression after renal transplantation [59]. mTOR, including mTORC1 and mTORC2, is abundant in podocyte and plays a role in development of podocyte damage [60]. Rheb activates mTOR signaling, but the activities of TSC1 and TSC2 complexes decrease in mTOR activation. TSC1/TSC2 is inhibited by phosphoinositide 3-kinase (PI3K) and is upregulated by 5′-adenosine monophosphate-activated protein kinase (AMPK) [61]. Epidermal growth factor activates the protein kinase B–PI3K pathway and abundant nutrients including HG and amino acid inhibit AMPK activation [62]. HG-induced epidermal growth factor signaling activation and AMPK inactivation propagate mTOR signaling, subsequently suppressing autophagy in early diabetic nephropathy; by contrast, mTOR inhibition enhances autophagy and prevents diabetic nephropathy [63]. However, chronic mTOR inhibition also induced podocyte injury in diabetic models and in kidney transplant recipients [64,65]. Taken together, a balance of mTOR signaling might be important for maintenance of podocyte integrity and development of podocyte damage [9]. A more specific Understanding of a mTOR pathway in podocyte might be applicable to therapeutic agents in glomerular diseases.
Conclusion
The effect of immunosuppressive agents in glomerular diseases is attributed to immune-dependent mechanisms. However, recent studies demonstrated that the antiproteinuric effect of these agents might be associated not only with their immunosuppressive action, but also via a direct effect on podocytes. Elucidation of direct target of the currently available immunosuppressive agents in podocytes might be helpful to search for new drugs without undesirable adverse reactions associated with immunosuppression.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









