A rare case of hyperoxaluria presenting with acute liver injury and stone-free kidney injury
Article information
Abstract
A 49-year-old woman visited the clinic because of acute hepatitis and acute kidney injury with decreased urine output presenting microscopic hematuria and proteinuria. An abdominal computed tomography revealed a localized, hypoattenuated lesion in a hepatic lateral segment, and kidney biopsy showed oxalate crystal deposition with tubular necrosis. In addition, the patient׳s 24-hour urinary excretion of oxalate was increased. Her kidney and liver injury improved after sessions of hemodialysis, and urinary oxalate excretion was normalized. Major mutations in primary hyperoxaluria have not been proven. A full sequencing of target genes may be helpful to diagnose a rare form of primary hyperoxaluria.
Introduction
Although acute kidney injury (AKI) due to intratubular crystal deposition can often occur in various clinical situations, oxalate nephropathy is an unusual cause of acute or chronic renal failure. Hyperoxaluria can develop in patients with inborn disorders of oxalate metabolism (primary hyperoxaluria) or secondary to other diseases (secondary hyperoxaluria) [1,2]. Primary hyperoxaluria is a rare autosomal recessive inherited metabolic disease. In primary hyperoxaluria, oxalate is overproduced, and nephrolithiasis repetitively occurs. Repetitive nephrocalcinosis can eventually cause end-stage renal disease [1–4]. Liver enzymes that specifically act on oxalate metabolism include alanine-glyoxylate aminotransferase (AGT), glyoxylate reductase-hydroxypyruvate reductase (GRHPR), and 4-hydroxy-2-oxoglutaratealdolase (HOGA) [1]. Secondary hyperoxaluria can occur through several pathways, including overingestion of oxalate or oxalate precursors and increased absorption into the body through the intestinal tract (e.g., ethylene glycol, inflammatory bowel disease, or pancreatic insufficiency) [2,5,6].
We report a patient with hyperoxaluria induced AKI and acute hepatitis.
Case report
A 49-year-old woman visited our hospital because of right upper quadrant discomfort and decreased urine output for 1 week. She denied having any previous medical problems, use of regular medications, or intake of unusual foods or beverages, with the exception of sorghum-derived Chinese liquor 1 week before the hospital visit. On admission, she had a blood pressure of 150/80 mmHg, heart rate of 88 beats/min, body temperature of 36.9°C, and respiratory rate of 20 breaths/min. Laboratory values on admission to the hospital were as follows: white blood cell count, 8,400/mm3 with 81.1% of segmental neutrophils; hemoglobin, 10.1 g/dL; sodium, 129 mEq/L; potassium, 4.0 mEq/L; chloride, 101 mEq/L; total carbon dioxide, 17 mEq/L (anion gap 11); calcium, 8.1 mg/dL; phosphorus, 8.6 mg/dL; uric acid, 6.0 mg/dL; blood urea nitrogen, 81 mg/dL; serum creatinine, 11.4 mg/dL; serum glutamic-oxalocetic transaminase, 695 U/L; serum glutamic pyruvic transaminase, 1,181 U/L; alkaline phosphatase, 117 U/L; gamma-GT, 111 IU/L; total bilirubin, 1.6 mg/dL; albumin, 2.9 g/dL; partial thromboplastin time, 35.0 seconds; prothrombin time (international normalized ratio), 0.97. The arterial blood gas showed the following: pH 7.383; pco2, 27.1 mmHg; po2, 84.7 mmHg; bicarbonate, 15.8 mmol/L.
Hepatitis A, B, and C serologies were negative. The abdominal ultrasonography showed relatively homogeneous echogenicity of the liver and no definite biliary dilatation. However, a nonenhanced abdominal computed tomography (CT) showed a localized, hypoattenuated lesion in the hepatic lateral segment suggesting acute hepatitis (Fig. 1A1).

Radiologic and pathologic findings. (A) Liver CT scan for the evaluation of liver function test (LFT) abnormality: liver CT scan at the time of admission shows a localized hypoattenuated area in the lateral segment of the liver (arrow; A1). CT scan after discharge shows the improvement of the lesion (arrow; A2: nonenhanced image, A3: enhanced image). (B) Kidney biopsy results: No cellular crescents and no evidence of focal sclerosis of the glomeruli are evident (B1, PAS,×100). Tubules focally reveal oxalate crystal deposits (arrow)(B2, PAS,×200) with necrosis (B3, PAS,×200). The interstitium exhibits focal moderate inflammatory cell infiltrates and fibrosis (B4, H&E,×200). CT, computed tomography; H&E, hematoxylin and eosin; PAS, periodic acid-Schiff.
The initial urinalysis showed 3+ protein, 10–19 red cells/high power field (HPF), and 1–4 white cells/HPF, and there were no evident crystals. The serum C3 and C4 levels were within normal limits. The antinuclear antibody test and antineutrophil cytoplasmic antibody were also negative. Because the patient had a kidney injury presenting as microscopic hematuria and proteinuria, we performed a kidney biopsy on the 5th day of admission to rule out glomerulonephritis. The histopathological findings revealed focal oxalate crystal deposition with tubule necrosis on light microscopy (Fig. 1B). Under electron microscopic examination, the thickness of the glomerular basement membrane was normal with partial wrinkling, and there were no electron-dense deposits. Immunofluorescence microscopy revealed no evidence of immune complexes or autoantibody deposition.
Because urine output has been reduced and azotemia has progressed, hemodialysis was started before the onset of symptoms due to AKI or signs of renal failure. Her renal function and urine output rapidly improved after several sessions of hemodialysis in the first 10 days of hospitalization (Fig. 2).
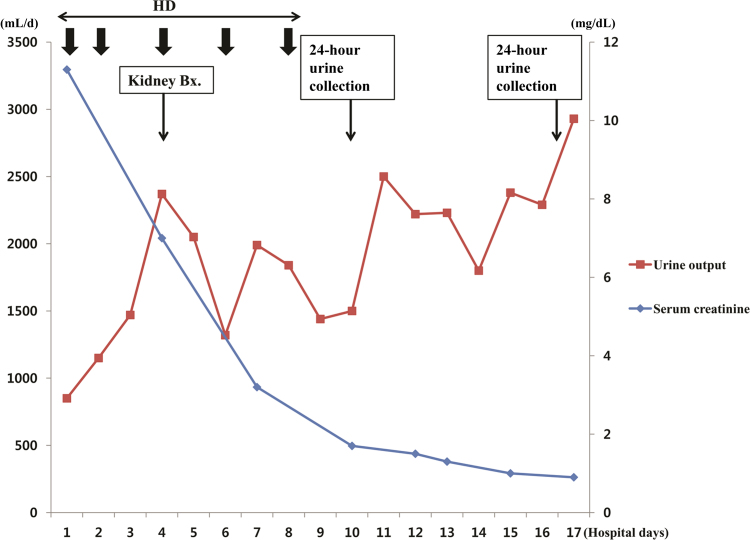
Changes in serum creatinine level and daily urine output throughout the hospital course. Bx., biopsy; HD, hemodialysis.
An analysis of the patient’s 24-hour urine collection taken on the 10th day of admission showed increased oxalate excretion [oxalate 68 mg/24 h (reference: 4–31 mg/24 h), citrate 63 mg/24 h (reference: ≥150 mg/24 h), calcium 66 mg/24 h (reference: 70–180 mg/24 h), and creatinine 0.9 g/24 h (reference: 0.8–2.0 g/24 h)] (Table 1). On the 17th day of admission, a 24-hour urine was collected again, and the laboratory values were as follows: oxalate 20 mg/24 h, Ca 79.1 mg/24 h, and citrate 542 mg/24 h. Because the possibility of secondary hyperoxaluria was low, a genetic analysis for major mutations associated with primary hyperoxaluria types 1 and 2 was performed. However, there were no meaningful mutations (Table 2).

Laboratory findings for the 24-hour urine collection throughout the clinical course

Gene sequencing profile of AGXT and GRPHPR
The patient was discharged 18 days after admission, and visited the outpatient clinic at 1 week and 5 weeks after discharge. Laboratory tests (liver panel, renal panel, urine analysis, and 24-hour urine oxalate excretion) were also repeated. All laboratory values continued to improve, and the 24-hour urine oxalate excretion was 20 and 27 mg/24 h at 1 and 4 weeks after the initial 24-hour urine collection, respectively. After renal function had normalized, an enhanced abdominal CT scan was performed and showed an improvement in the localized hypoattenuated lesion in the hepatic lateral segment (Figs. 1A2 and 1A3).
Discussion
Oxalate is the end product of ascorbic acid and glyoxalate in catabolic metabolism in humans [1]. Excretion through the kidney is the only way to eliminate oxalate because oxalate is not metabolized in the human body. As excretion of oxalate through the kidneys increases, oxalate crystals are deposited in the tubular lumen and renal pelvis. This deposition leads to impaired tubular epithelium, obstruction of the tubular lumen, and recurrent urolithiasis, resulting in a deceased glomerular filtration rate [7].
In this patient, oxalate deposition was found on the kidney biopsy, and increased oxalate excretion was confirmed by the 24-hour urine collection. However, the patient had no history of urinary calculi, and no specific findings of nephrolithiasis were found by abdominal CT and ultrasonography. With regard to secondary hyperoxaluria, the patient had no dietary habits consistent with increased ingestion of oxalate or oxalate precursors and showed no symptoms suggestive of gastrointestinal diseases causing oxalosis [2,5]. Moreover, symptoms suggestive of ethylene glycol ingestion were not seen. In addition, the patient was not an alcoholic, and the serum anion gap was not elevated [6]. Collectively, these findings suggested the possibility that primary hyperoxaluria led to oxalate deposition. A genetic analysis for major mutations that cause primary hyperoxaluria types 1 and 2 was then performed, but no major mutations were detected [8–12].
However, the potential risk of primary hyperoxaluria cannot be excluded. First, findings suggestive of secondary hyperoxaluria were not found in the patient’s history. Second, the possibility of an unusual phenotype of primary hyperoxaluria with a later progression than the typical clinical presentation of primary hyperoxaluria remains [13].
The point that we should focus on is that the patient presented with AKI related to oxalate crystal deposition and acute hepatitis. One hypothesis to explain the present case is that acute liver injury resulted in a deficiency of the liver-specific enzymes related to glyoxylate-metabolism (e.g., AGT, GRHPR), leading to increased oxalate formation. Then, oxalate deposition resulted in renal tubule obstruction and AKI. Notably, the 24-hour urinary excretion of oxalate improved after normalization of the liver enzymes. Many cases of hepatitis accompanied by AKI have been reported. However, to our knowledge, no previous reports have described metabolic disorders controlled by liver enzymes associated with preceding acute liver injury, especially in adults, although Reye-like syndrome, an inherited metabolic disease, can present with acute onset liver failure [14]. Additional studies on the temporary dysfunction of liver-specific enzymes due to hepatitis as a cause of increased oxalate formation could be of broader interest.
The progress of the patient will be observed by monitoring renal function, signs of urolithiasis, and the urinary oxalate excretion level because the cause of hyperoxaluria was not well defined. Moreover, we are planning to perform full sequencing of the candidate genes for primary heperoxaluria if a similar event occurs in the future. In some cases that have been reported, no causative mutations were detected by genetic analysis, despite the typical clinical manifestations of primary hyperoxaluria. This result occurred because mutations exist in promoters or other regulatory sequences or mutations that have not yet been revealed exist, resulting in disrupted metabolism. In the case of recurrence, we can also consider performing a liver biopsy to evaluate the expression of AGT, GRHPR, and HOGA to prove abnormal oxalate metabolism [15].
If crystal deposition is found on the kidney biopsy without any evidence of other renal diseases, oxalosis should be considered, and appropriate evaluations, including urine oxalate excretion, should be performed. If secondary hyperoxaluria can be excluded, primary hyperoxaluria should be considered as a possibility, and evaluations, including genetic analysis, should be performed.
Conflict of interest
The authors have no financial conflict of interest.