



Introduction
Fabry disease is an X-linked lysosomal storage disease caused by mutations in the gene encoding the enzyme ╬▒-galactosidase A (╬▒-GLA gene) [1]. Deficient or absent activity of ╬▒-GLA causes progressive accumulation of undegraded glycosphingolipid products, predominantly globotriaosylceramide (GL3), within lysosomes in the cells of many tissues, including the kidney [2,3]. Fabry disease causes progressive cardiovascular, neurological, and renal complications, all potentially fatal [4ŌĆō6]. Renal manifestation (proteinuria and progressive renal failure) occurs early in the course of Fabry disease and tends to be more severe in males than in females [7ŌĆō9].
Enzyme replacement therapy (ERT) has been reported to slow the deterioration of renal function in patients with Fabry nephropathy [10]. However, ERT was shown in previous studies to have a limited effect on progressive renal damage when initiated at late stages [11,12]. Although kidney biopsy is one of the best methods to investigate the effect of ERT on Fabry nephropathy, the procedure is not easily performed due to its invasiveness. However, previous kidney biopsy studies have shown that long-term ERT could induce and sustain clearance of GL3 deposits from various types of kidney cells [13,14]. However, de novo or persistent podocyte GL3 accumulation and progressive foot process effacement (FPE) were reported in young patients with Fabry disease despite 3 to 5 years of ERT [15]. The authors also showed that podocyte accumulation of GL3 and FPE might be present in patients with Fabry disease without clinically evident kidney involvement (normal glomerular filtration rate [GFR] and normoalbuminuria) [15].
Due to the heterogeneous kidney biopsy findings in patients with Fabry disease, the effects of ERT on kidney pathology in patients with Fabry disease who had received ERT were investigated in the present study. In addition, the pathologic kidney findings in normoalbuminuric patients with Fabry disease who were naïve to ERT were evaluated.
Methods
A total of 15 patients (3 males and 12 females) with Fabry disease who underwent kidney biopsy at Pusan National University Yangsan Hospital were retrospectively investigated in this study. Fabry disease was diagnosed based on clinical manifestations and ╬▒-GLA gene mutation and/or ╬▒-GLA analysis findings. Among the cases, nine were the classical type and six were the late-onset type. The patients were divided into two groups. Group 1 (n = 9, patients No. 1ŌĆō9) had received ERT (agalsidase ╬▒ or ╬▓) at the time of kidney biopsy, and group 2 (n = 6, patients No. 10ŌĆō15) had not received ERT at the time of kidney biopsy. The drug history for angiotensin-converting enzyme inhibitor (ACEI) or angiotensin receptor blocker (ARB), which can affect the degree of proteinuria, was also investigated.
All research and data collection processes were conducted in accordance with the Declaration of Helsinki and current ethical guidelines. This study protocol was approved by the Institutional Review Board (IRB) of Pusan National University Yangsan Hospital (No. 05-2020-226). The need for informed consent was waived by the IRB due to the retrospective nature of the analysis and only anonymized information contained in medical charts and records was used.
The urinary albumin-to-creatinine ratio and protein-to-creatinine ratio were measured in morning urine samples (means of two consecutive samples). An albumin-to-creatinine ratio of <30 mg/g was considered to indicate normoalbuminuria. Albumin-to-creatinine ratios of 30ŌĆō300 mg/g and >300 mg/g were considered to indicate microalbuminuria and macroalbuminuria, respectively. Renal function was measured using the estimated GFR (eGFR). The eGFR was calculated from the serum creatinine level using the Chronic Kidney Disease-Epidemiology formula for adults [16] or the Schwartz formula for children [17]. Both eGFR values have been validated in patients with Fabry disease [18].
The kidney biopsy findings were examined by an experienced nephropathologist. For standard light microscopy, the biopsy tissue was stained with hematoxylin and eosin, periodic acid-Schiff, and silver methenamine. The biopsy specimens were evaluated using light microscopy based on changes in the glomeruli (global or focal glomerulosclerosis), tubules (tubular atrophy), interstitium (interstitial fibrosis), and vessels (hyaline change in the media). GL3 deposits in podocytes, mesangial cells, glomerular endothelial cells, and tubular epithelial cells were scored as present or absent on electron microscopy (EM). The degree of segmental FPE was evaluated using EM and expressed as the percentage of the length with FPE out of the entire length of the capillary loops.
Results
Table 1 shows the clinical features of the 15 patients with Fabry disease (three males and 12 females). All patients underwent renal biopsy while receiving ERT (group 1, patients No. 1ŌĆō9) or before ERT initiation (group 2, patients No. 10ŌĆō15). The age range was 19 to 58 years in group 1 and 11 to 66 years in group 2. Most patients had acroparesthesia, vortex keratopathy, and angiokeratoma of various degrees. Most patients in group 1 showed improved clinical symptoms, such as acroparesthesia (pain score from 2.8 ┬▒ 1.9 to 0.7 ┬▒ 0.7) while receiving ERT.
The baseline characteristics of the 15 patients with Fabry disease are summarized in Table 2. None of the 15 patients had diabetes mellitus. Two patients (No. 1 and 5) had hypertension. The ERT duration was 1.2 to 8.0 years in group 1. ACEI or ARB (duration, 1.0ŌĆō4.5 years) was used in six of the nine patients in group 1 at the time of kidney biopsy. Conversely, neither of these drugs were used in any patient in group 2 at the time of renal biopsy. The plasma globotriaosylsphingosine (lyso-Gb3) levels were 10.4 ┬▒ 14.6 ng/mL in group 1 and 29.8 ┬▒ 54.1 ng/mL in group 2. In group 1, the mean albumin-to-creatinine ratio was 125.9 ┬▒ 179.3 mg/g (range, 5.4ŌĆō530.0 mg/g). Five of the nine patients (patients No. 1, 2, 3, 7, and 8) showed normoalbuminuria. Three patients (patients No. 4, 5, and 6) showed microalbuminuria. Only one patient (patient No. 9) showed macroalbuminuria. In group 2, the mean albumin-to-creatinine ratio was 13.8 ┬▒ 5.8 mg/g (range, 6.6ŌĆō21.5 mg/g). All patients in group 2 showed normoalbuminuria. The mean protein-to-creatinine ratio was 242.3 ┬▒ 254.7 mg/g (range, 43.2ŌĆō731.0 mg/g) in group 1 and 81.1 ┬▒ 24.4 (range, 55.0ŌĆō125.0 mg/g) in group 2. The mean eGFR was 112.9 ┬▒ 20.1 mL/min/1.73 m2 (range, 80ŌĆō137 mL/min/1.73 m2) in group 1 and 111.0 ┬▒ 22.4 mL/min/1.73 m2 (range, 82ŌĆō137 mL/min/1.73 m2) in group 2.
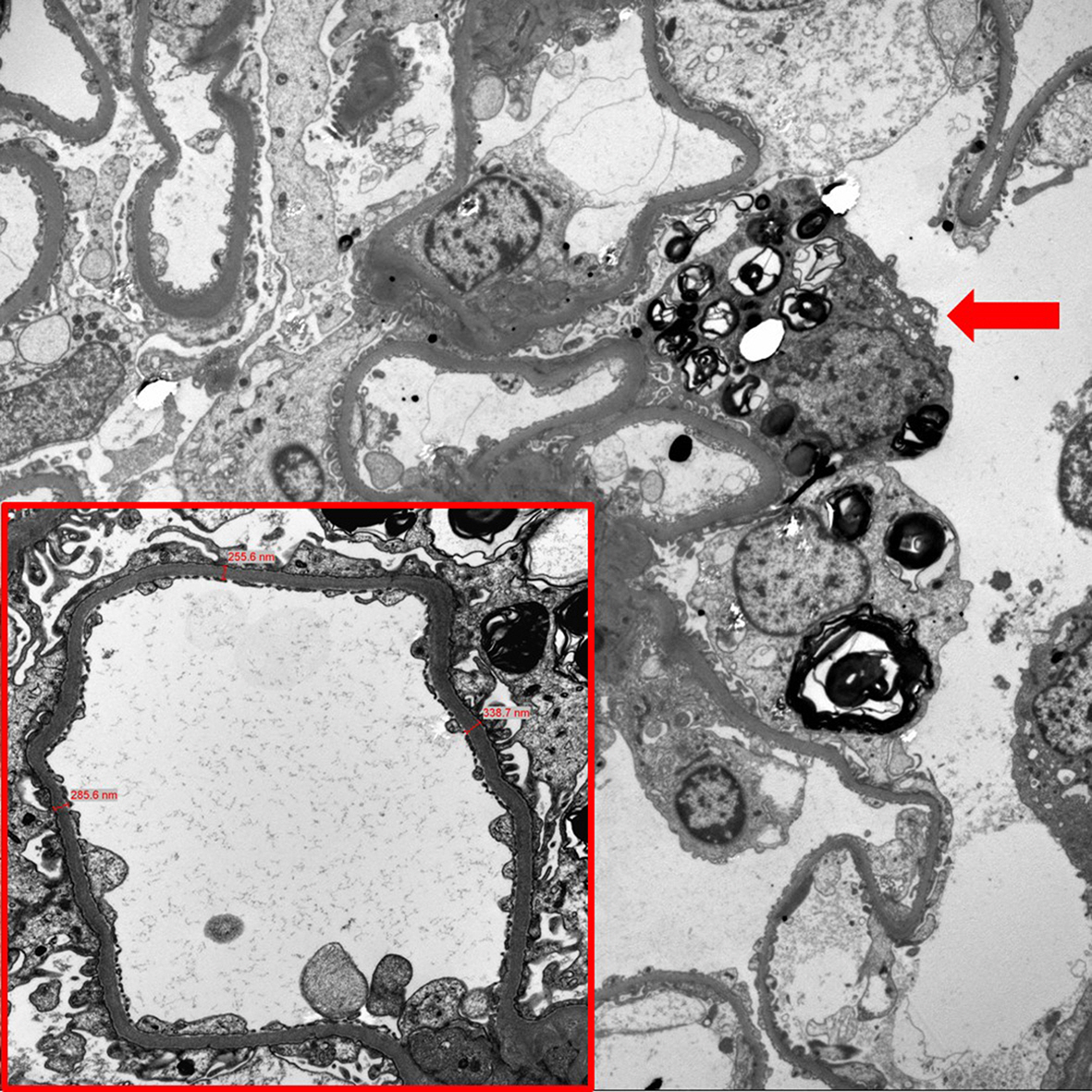
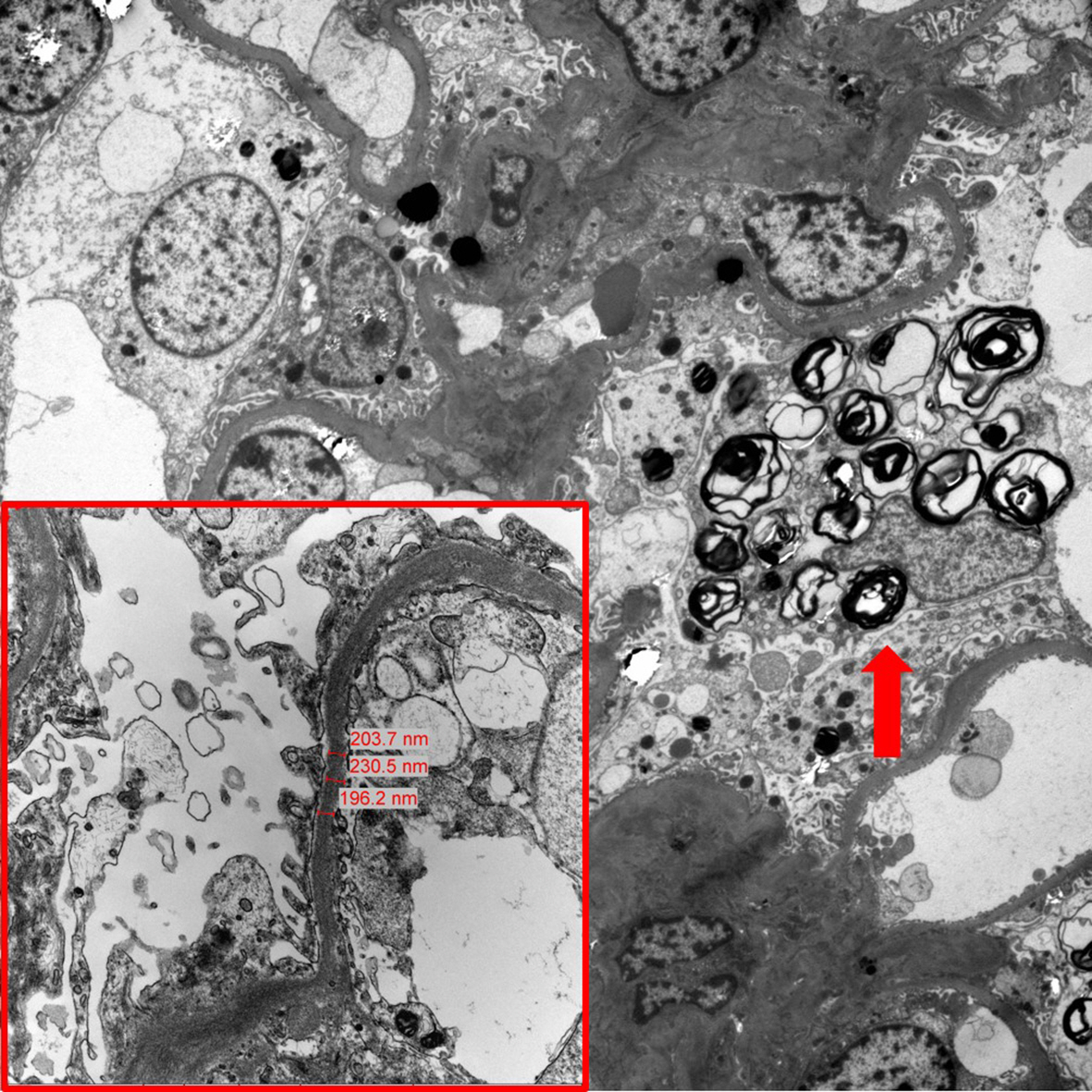
Light microscopy showed glomerular, tubular, interstitial, and vascular changes in some patients, either alone or in combination (Table 3 and Fig. 1) as follows: global glomerular sclerosis, 5 of 9 patients in group 1 and 2 of 6 patients in group 2; segmental glomerular sclerosis, 3 of 9 patients in group 1 and 2 of 6 patients in group 2; tubular atrophy, 1 of 9 patients in group 1 and 1 of 6 patients in group 2; interstitial fibrosis, 4 of 9 patients in group 1 and 3 of 6 patients in group 2; and vasculopathy, 1 of 9 patients in group 1 and 2 of 6 patients in group 2. EM showed segmental FPE and GL3 deposits in the podocytes, mesangium, endothelium, and tubular epithelium in most patients in groups 1 and 2 (Table 4) as follows: segmental FPE, 7 of 9 patients in group 1 and 6 of 6 patients in group 2; podocyte GL3 deposits, 7 of 9 patients in group 1 and 6 of 6 patients in group 2; mesangial GL3 deposits, 3 of 9 patients in group 1 and 5 of 6 patients in group 2; endothelial GL3 deposits, 2 of 9 patients in group 1 and 4 of 6 patients in group 2; and tubular epithelial GL3 deposits, 3 of 9 patients in group 1 and 5 of 6 patients in group 2. Notably, patients No. 1 and 7 in group 1 showed no FPE or GL3 deposits (Fig. 2). The seven remaining patients (patients No. 2, 3, 4, 5, 6, 8, and 9) in group 1 showed segmental FPE and podocyte GL3 deposits of various degrees despite ERT. Conversely, all patients in group 2 (patients No. 10ŌĆō15) showed segmental FPE and podocyte GL3 deposits, indicating that segmental FPE may be present in patients with Fabry disease with no clinical signs of renal involvement (Fig. 3). Most patients in group 2 also showed GL3 deposits in the mesangium, endothelium, or tubular epithelium.
Discussion
Progressive nephropathy (Fabry nephropathy) is the main feature of Fabry disease [8]. In the kidney, progressive deposits of GL3 affect all types of kidney cells, including tubular, glomerular, endothelial, and vascular smooth muscle cells [19ŌĆō21]. In untreated patients with classical mutations, Fabry nephropathy leads to end-stage renal disease from the third to the fifth decade of life [8]. Approximately 30% to 35% of females with Fabry disease have proteinuria and 1% to 4% have end-stage renal disease [9]. Therefore, kidney biopsy in Fabry nephropathy could be a useful tool for investigating its mechanism, progression, and treatment. However, studies in which the pathologic lesions of Fabry nephropathy have been investigated are limited.
In the present study, typical findings of GL3 accumulation were observed in the podocytes, mesangium, endothelium, and tubular epithelium in the majority of patients with Fabry disease. In group 1, seven of the nine patients (patients No. 2, 3, 4, 5, 6, 8, and 9) showed persistent GL3 deposits in the podocytes and segmental FPE despite 1.2 to 4.0 years of ERT. Only two patients (patients No. 1 and 7) showed no GL3 deposits and segmental FPE with 8.0 and 5.2 years of ERT, respectively. At the time of kidney biopsy, the average plasma lyso-Gb3 level in these two patients was 3.6 ng/mL, which was lower than the lyso-Gb3 level of 13.4 ng/mL in the other seven patients. Although the ERT duration was relatively shorter in the seven patients (3.4 years) than in the two patients (6.6 years), these observations indicated that ERT might not prevent or clear the GL3 deposits in the kidneys of some patients with Fabry disease. Conversely, in the present study, patients in group 1 showed lower rates of segmental FPE and GL3 deposits in podocytes, mesangium, endothelium, and tubules than patients in group 2, indicating this observation may be due to the effects of ERT. Regarding the effect of ERT on GL3 deposits in the kidney, inconclusive results have been shown in previous studies. Skrunes et al. [22] showed in 12 patients with Fabry disease that long-term ERT for up to 14 years can result in the reduction of podocyte GL3 deposits, which correlated with the cumulative ERT dose. However, in the present study, limited clearing of arterial GL3 deposits raised concerns regarding the long-term vascular effects of ERT [22]. Lubanda et al. [23] showed that a lower dose of ERT might be sufficient in some but not all patients with Fabry disease to maintain GL3 clearance in various kidney cell types. T├Ėndel et al. [15] reported the de novo appearance of GL3 deposits and segmental FPE in patients with Fabry disease who showed no FPE at baseline kidney biopsy after 3 years of ERT. The authors also reported persistent GL3 deposits and progression of segmental FPE in two patients with Fabry disease treated with ERT for 5 years [15]. These findings, including the results in the present study, indicate that multifactorial mechanisms might be involved in progressive Fabry nephropathy in addition to GL3 accumulation in the kidney cells.
The most important finding observed in the present study was the segmental FPE in the patients with Fabry disease who had normoalbuminuria and were na├»ve to ERT and ACEI/ARB (patients No. 10ŌĆō15). To date, early segmental FPE in normoalbuminuric patients with Fabry disease has been reported in only a few studies [15,24,25]. Podocytes are highly specialized epithelial cells that cover the glomerular basement membrane (GBM) with numerous interdigitating foot processes [26]. The GBM and podocytes are key components of the glomerular filtration barrier. Proteinuria is associated with significant changes in podocyte architecture, which include loss of the podocyte FPE [26]. FPE is regarded as a stereotypical reaction of podocytes to injury or damage. Although whether FPE in Fabry nephropathy is secondary to GL3 deposits or associated with mechanisms other than GL3 deposition is unknown, a strong association between podocyte GL3 accumulation and FPE was demonstrated in a previous study. Najafian et al. [27] recently showed that podocyte GL3 volume was associated with podocyte injury and loss, evidenced by increased foot process width and decreased podocyte number density. The authors also showed that increased podocyte GL3 volume and foot process width were associated with increased urinary protein excretion as well as decreased GFR. Finally, they suggested that podocyte injury plays an important role in the progression of Fabry nephropathy and a need for ERT before critical podocyte loss occurs. The results of the present study also indicated that FPE might be an early sign of Fabry nephropathy, preceding the onset of overt albuminuria. Because overt albuminuria has been reported a strong predictor and to indicate the irreversible stage of progressive renal dysfunction in proteinuric renal disease as well as Fabry nephropathy, these early morphological changes could be clinically significant as a prealbuminuric marker of Fabry nephropathy. However, the results need to be interpreted cautiously because biopsy sampling errors may be present; possibly segmental FPE and GL3 deposition could be missed during biopsy sampling. Thus, further kidney biopsy studies that include a large number of patients with Fabry disease are needed to confirm the present and previous study results.
This study had several limitations. First, due to the retrospective design, comparing the kidney biopsy findings before and after ERT in the same patient was not possible. Thus, longitudinal studies in which the kidney biopsy findings are compared before and after ERT in each patient are necessary to determine if the ERT could lead to amelioration of Fabry nephropathy. Second, in the female patients with Fabry disease, the clinical characteristics are heterogeneous due to skewed X-chromosome inactivation. In the present study, most patients were females. The situation for females would be more complex because their podocyte involvement and injury are affected by mosaicism resulting from skewed X-chromosome inactivation. Therefore, in the future, performing X-chromosome inactivation analysis will help to understand the association between the X-chromosome inactivation and degree of kidney injury in female patients with Fabry disease.
Despite the small number of patients with Fabry disease included in the study, important clinical implications were determined. The study results showed that GL3 deposits in the kidney and segmental FPE may be present in patients with Fabry disease experiencing improved clinical symptoms while receiving ERT. This observation indicates the suboptimal effect of the ERT regimen in these patients. Although the only clearance of GL3 deposits confers unequivocal long-term prevention or stabilization of Fabry nephropathy [15], the study results showed that ERT response may be asynchronous between clinical symptoms, such as acroparesthesia and Fabry nephropathy. Thus, the important role of kidney biopsy in the assessment of the response to ERT was proven in this study. In addition, GL3 deposits in the kidney and segmental FPE in normoalbuminuric patients with Fabry disease were observed. To date, albuminuria and GFR reduction are the only clinical markers of renal dysfunction in Fabry disease [8]. Thus, the initiation of ERT is often delayed until proteinuria or GFR reduction occurs, during which the reversibility of renal damage is already difficult to achieve [8]. Generally, kidney biopsy is not recommended for kidney disease that does not show proteinuria [28]. However, the results of the present study showed that kidney biopsy is essential even in patients with Fabry disease who present with normoalbuminuria to confirm kidney involvement of Fabry disease and initiate timely ERT intervention.
In conclusion, segmental FPE and GL3 deposits can be persistent in Fabry nephropathy despite ERT, indicating the response to ERT could be asynchronous between clinical symptoms and Fabry nephropathy. In addition, segmental FPE and GL3 deposits were observed in various kidney cell types in normoalbuminuric patients with Fabry disease, indicating that albuminuria is not sufficiently sensitive to detect early kidney injury in Fabry nephropathy. These results indicate that kidney biopsies at baseline and follow-up evaluation of Fabry nephropathy are essential for timely ERT initiation and ERT response assessment.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









