


Introduction
Regardless of the cause of the initial injury, chronic kidney disease (CKD) frequently progresses to end-stage renal disease with the pathogenesis of fibrosis and complete destruction of functional kidney tissues. CKD has become a major public health concern worldwide as the incidence continues to rise and portends high rates of morbidity and mortality [1]. The hallmark of progressive CKD is the development of renal fibrosis that is thought to be the final common mechanism leading to end-stage renal disease [2], [3], [4]. In general, fibrosis is characterized by the continuous production and progressive accumulation of extracellular matrix (ECM) proteins, including collagen and fibronectin, in the tissues. Renal fibrosis shows significant correlation with deterioration of kidney function [4], [5]. The growing body of evidence demonstrates that transforming growth factor-β1 (TGF-β1) plays a pivotal role in the pathogenesis of renal fibrosis associated with progressive kidney diseases [6], [7]. Therefore, improved and more effective therapies with direct antifibrotic effects are highly potent therapeutic strategies for attenuation or prevention of progressive CKD.
There are three mammalian isoforms, TGF-β1, TGF-β2, and TGF-β3, of which TGF-β1 represents the predominant isoform and the prototype member of the TGF-β superfamily of multifunctional cytokines. TGF-β1 has been shown to be the key regulator of a variety of cellular functions such as cell growth, cellular differentiation, apoptosis, and wound healing, and is a potent inducer of ECM synthesis [8], [9]. In response to tissue injury, upregulation of TGF-β1 expression and consistent activation is a common finding in the pathogenesis of renal fibrosis seen in virtually every type of CKD [6], [10], [11]. In the acute phase, however, TGF-β1 also triggers cytoprotective effects to mitigate tissue injury through enhancing wound repair and tissue regeneration, as well as anti-inflammatory effects [11], [12], [13], [14], [15], [16].
Thus, it seems that TGF-β1 plays a paradoxically dual role in tissue injury response, suggesting that simply inhibiting the function of TGF-β1 receptors or TGF-β1 may not be an appropriate strategy for therapeutic interventions in CKD. In this context, a more detailed understanding of the cellular and molecular mechanisms of TGF-β1 actions will not only provide a more comprehensive knowledge of the pathogenic mechanisms in CKD, but may also guide the development of therapeutic strategies specifically targeting the signaling pathway responsible for the deleterious effects of TGF-β1.
TGF-β receptors
TGF-β1 signals are transmitted through transmembrane serine/threonine kinase receptors, type I (TβRI) and type II (TβRII), to activate intracellular downstream signaling pathways [17]. In the absence of the ligand TGF-β1, TβRI and TβRII exist as homodimers at the cell surface. Upon ligand stimulation, TGF-β1 binds to TβRII, and in turn TβRI and TβRII form heterotetrameric complexes. Since TβRII dimer is a constitutively active kinase receptor, upon ligand binding it phosphorylates serine/threonine residues in the cytoplasmic GS domain of TβRI. However, TβRII signaling in the absence of TβRI has not been reported. The phosphorylation of serine/threonine residues in the GS domain activates TβRI, and this is followed by activation of a number of intracellular signaling molecules in a cell-specific and context-specific manner to mediate the diverse biological functions of TGF-β1.
Although TGF-β1 binds efficiently to TβRI–TβRII complexes, TGF-β type III receptor (TβRIII), also known as betaglycan, which lacks a signaling domain, serves as a co-receptor to promote the binding of TGF-β ligands to TβRII in certain cells [18]. This function of TβRIII appears to be particularly important for TGF-β2. In contrast to TGF-β1 and TGF-β3, affinity of TGF-β2 for TβRII is much weaker and requires betaglycan for high-affinity binding to TβRII [19].
Signaling pathways induced by TGF-β1
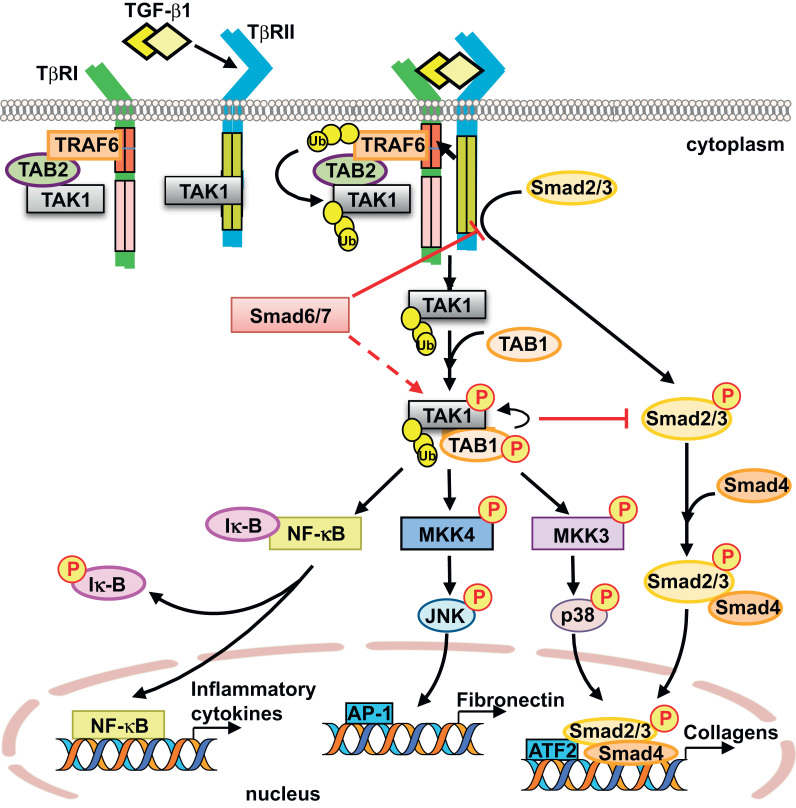
A comprehensive overview of TGF-β-activated Smad-dependent and Smad-independent signaling pathways is shown in Fig. 1. The first member of the Smad family, Mad [mothers against dpp (decapentaplegic)], was identified in a genetic screen in Drosophila
[17], [20], and followed by cloning of sma-2, sma-3 and sma-4 (Small body size) in Caenorhabditis elegans
[21], [22].
Phosphorylation in the GS domain of TβRI resulting in its receptor kinase activity recruits and activates receptor-regulated Smads (R-Smads). In addition to the phosphorylation in the GS domain [23], the nine-amino-acid L45 loop [24] of TβRI is thought to be crucial for its interaction with R-Smads. The recruitment of R-Smads to the receptor complex is mediated by auxiliary proteins, such as Smad anchor for receptor activation (SARA) [25]. R-Smads, Smad2 and Smad3, are phosphorylated by kinase activity of TβRI and rapidly dissociate from TβRI. Subsequently, the phosphorylated R-Smads interact to form complexes with the common mediator (Co-Smad) Smad4, leading to nuclear translocation and transcriptional activity [26]. Transcriptional activation of Smad complexes leads to cooperate with other co-activators, such as p300 and CREB-binding protein (CBP), which possess histone acetyl transferase activity [27]. On the other hand, the inhibitory Smads (I-Smads), Smad6 and Smad7, inhibit TGF-β signaling through binding of their MAD homology (MH) 2 domains to TβRI, thus preventing the recruitment and phosphorylation of R-Smad [17], [28].
The Smad signaling pathway is widely accepted as a canonical pathway induced by TGF-β1 [29], and the role of Smads in kidney diseases has been a topic of several previous reviews [30], [31]. Nevertheless, it has become quite evident that the Smad signaling pathway does not explain all of the diverse actions of TGF-β1. A large body of evidence demonstrates that TGF-β1 also induces the activation of various Smad-independent signaling pathways, with or without direct crosstalk with the Smad [32], [33].
The Smad-independent TGF-β signaling pathways, as illustrated in Fig. 1, include the mitogen-activated protein kinases (MAPKs), namely extracellular signal-regulated kinases 1/2 [34], [35], c-Jun N-terminal kinase (JNK) [36], [37], [38], and p38 MAPK [39], [40], [41], [42], phosphatidylinositol-3-kinase (PI3K)/AKT [43], [44], [45], [46], Rho-like GTPases (RhoA) [47], [48], and protein phosphatase 2A (PP2A) [49]. Recent studies have demonstrated the role of p38 MAPK signaling pathway in the development of glomerular and tubulointerstitial fibrosis [50], [51] in animal models and in human kidney disease such as diabetic nephropathy [50], [52]. We and others have demonstrated that TGF-β-activated kinase 1 (TAK1) is a major upstream signaling molecule in TGF-β1-induced type I collagen and fibronectin expression through activation of the MAPK kinase (MKK) 3–p38 and MKK4–JNK signaling cascades, respectively (Fig. 2) [53], [54], [55]. Here, we review recent progress toward understanding the molecular mechanisms of Smad-independent signaling pathway via TAK1 and its role in mediating the cellular effects of TGF-β1.
TAK1 in TGF-β signaling
TAK1, a serine/threonine kinase, was originally identified as a member of the MAPK kinase kinase (MAP3K) family, named as MAP3K7, and is rapidly activated by TGF-β1 [56], [57]. To date, TAK1 is the only MAP3K family member that has been directly implicated in TGF-β1 signaling. In addition to TGF-β1, TAK1 can also be activated by various stimuli including proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) [58] and interleukin-1 (IL-1) [59], lipopolysaccharides [60], and environmental stress [61]. Phosphorylation of Thr-187 and Ser-192 in the activation loop of TAK1 induces TAK1 activation [62], [63] and subsequently triggers the activation of several downstream signaling cascades, including MKK4/7–JNK, MKK3/6-p38 MAPK, and nuclear factor-kappa B (NF-κB)-inducing kinase-IκB kinase (Fig. 2) [58], [59], [60].
Recent investigations also indicate a role for TAK1 in the regulation of Smad function. TAK1 interacts with the MH2 domain in Smad proteins, via which TAK1 dramatically interferes with R-Smad transactivation and affects the subcellular distribution of Smad proteins [64]. Similarly, it has been demonstrated that IL-1β transiently induces the association between TAK1 and the MH2 domain of Smad3 and inhibits TGF-β-induced Smad3 signaling [65]. These studies indicate that TAK1 negatively regulates R-Smad activation through direct interaction. Furthermore, TAK1 is able to repress Smad activation through upregulation of inhibitory Smad7 expression [66]. Intriguingly, however, TAK1 can also enhance TGF-β1-induced Smad signaling. SnoN, an inhibitor of TGF-β signaling, recruits transcriptional repressor complex to block Smad-dependent transcriptional activation of TGF-β-responsive genes [67]. Following TGF-β stimulation, TAK1 interacts with and phosphorylates SnoN, resulting in rapid degradation of SnoN and thereby allowing the activation of TGF-β target genes by R-Smad [68].
There is also evidence to suggest a role of I-Smads in the regulation of TAK1 function. Smad6 and Smad7 have been shown to reversely inhibit TAK1 activation in certain cell types, such as rat pheochromocytoma PC12 cells [69] and mouse hybridoma MH60 cells [70]. Conversely, in human prostate cancer PC-3U cells, Smad7 may act as a scaffolding protein and facilitate the activation of the TAK1–MKK3–p38 signaling axis [71]. Thus, the reciprocal regulation between TAK1 and Smad proteins appears to be dependent on cell type and context.
In addition to the role of TAK1 in the regulation of Smad function, downstream targets of TAK1 such as p38 MAPK and ATF2 crosstalk with Smads to regulate expression of certain TGF-β1 target genes [39], [72], [73]. Collectively, these findings suggest that TAK1 might play a pivotal role in regulating TGF-β signaling.
Molecular mechanism of TAK1 activation
Requirement of TAK1-binding proteins
Among the MAP3K family members, TAK1 is unique in that its activation requires the formation of complexes with specific binding partner proteins known as TAK1-binding proteins 1, 2, and 3 (TAB1, TAB2, and TAB3) [74], [75], [76]. TAB1 and TAB2 are structurally unrelated TAK1-binding proteins, whereas TAB3 is a TAB2-related protein that shares 48% amino acid sequence identity with TAB2 [75], [76]. Studies in embryonic fibroblasts from Tab1-deficient mice have demonstrated that osmotic stress induces TAK1 activation in a TAB1-dependent fashion [77], and that TAB1 is also essential for TAK1 activity and necessary for TGF-β signal transduction [78]. In vivo studies also indicate that the TAK1–TAB1 complex plays a pivotal role in embryonic development and morphogenesis, as Tab1 deletion in mice is embryonically lethal and causes defects in the development of major organs including the heart and lung [78]. We have also shown that TAB1 is indispensable for TGF-β1-induced TAK1 activation in glomerular mesangial cells [79]. By contrast, TNF-α and IL-1-induced activation of TAK1 did not require TAB1 [77], but rather TAB2 and TAB3 function redundantly as mediators of TAK1 activation in TNF-α and IL-1 signaling transduction [75]. Thus, the requirement for these TAK1-binding proteins appears to be dependent on the stimuli.
Moreover, there is evidence that supports cell-type specificity in the involvement of the specific TAK1-binding proteins. For instance, TNF-α and IL-1 induced activation of TAK1 is entirely normal in Tab1-deficient mouse embryonic fibroblasts [77], indicating that TAB1 is not required, whereas in HeLa cells, TNF-α-induced TAK1 activation involves a signaling complex with TAB1 as well as TAB2 [63]. In addition, in skin epithelial cells, TAB1 but not TAB2 is essential for TAK1 activation [80]. Interestingly, in the intestinal epithelium, TAK1 activity appears to be regulated through two independent mechanisms, in which TAB1 regulates basal activity of TAK1, and TAB2 mediates stimulus-dependent TAK1 activation [80]. Moreover, ablation of Tab1 downregulates, but double ablation of Tab1 and Tab2 abolishes, in vivo epithelial TAK1 activity, and epithelial-specific Tab1 and Tab2 double-knockout but not Tab1 or Tab2 single-knockout mice phenocopy epithelium-specific Tak1 knockout mice [80].
Requirement of ubiquitin ligase TNF receptor-associated factor 6
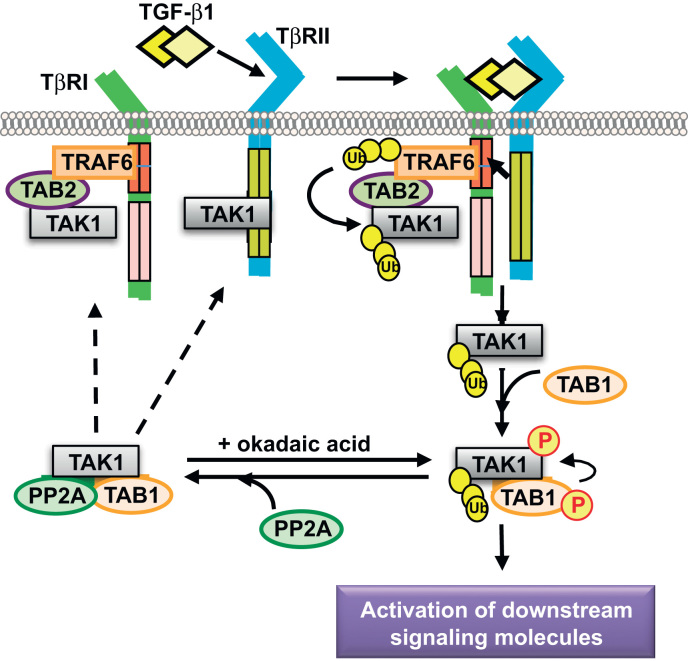
Despite the remarkable progress that has been made in unraveling the mechanism involving TGF-β-induced R-Smad activation, events that link the activation of non-Smad signaling molecules are less clearly understood. Recent evidence has demonstrated that the TAK1 activation mechanism is quite different from that of Smad2/3. Perhaps the most interesting difference is that TGF-β1-induced TAK1 activation occurs independently of TβRI kinase activity [79], [81], whereas activation of Smad2/3 involves recruitment and phosphorylation by TβRI and requires kinase activity of TβRI [23], [24]. Activated Smad2/3 are released from the receptor complex to interact with Smad4 to transmit TGF-β1 signals. By contrast, TAK1 has been shown to be stably associated with TβRI in the absence of ligand stimulation in glomerular mesangial cells [79]. Upon TGF-β1 stimulation, TAK1 is rapidly dissociated from the TβRI–TβRII complex, TAK1 is subsequently activated by its interaction with TAB1 and TAB1-mediated autophosphorylation, and TGF-β1-induced TAK1 activation occurs independently of receptor kinase activity of TβRI in glomerular mesangial cells [79]. Thus, although TAB1 is indispensable for TGF-β1-induced TAK1 activation in glomerular mesangial cells, TAB1 itself does not interact with TGF-β receptors and is not required for the TAK1–TβRI interaction.
However, the association of TAK1 with TβRI requires TAB2 and an additional adapter protein known as TNF receptor-associated factor 6 (TRAF6). The ubiquitin ligase (E3) TRAF6 is a member of a family of RING (really interesting new gene) domain ubiquitin ligases that catalyzes the synthesis of polyubiquitin chains linked through Lys-63 of ubiquitin. The highly conserved ubiquitin-binding zinc finger domain in TAB2 preferentially binds Lys-63-linked polyubiquitin chains on TRAF6 and facilitates TGF-β1-induced TAK1 activation [81], [82], [83]. TβRI contains a consensus binding site (basic residue-X-P-X-E-X-X aromatic/acidic residue) for TRAF6, by which TRAF6 physically interacts with TβRI and promotes Lys-63-dependent polyubiquitination of TAK1 at Lys-34 and subsequent TAK1 activation [81], [82], [83], [84]. However, several recent studies have claimed that Lys-158 on TAK1 instead of Lys-34 is the polyubiquitination target site of TRAF6 for TAK1 activation [85], [86], [87], although the reason for the discrepancy of polyubiquitin site on TAK1 remains unclear.
Nonetheless, there are notable differences in the mechanism of Smad2/3 and TAK1 activation. TβRI kinase activity is required for activation of the canonical Smad signaling pathway, whereas ubiquitin ligase activity of TRAF6 regulates the activation of TAK1 in a receptor kinase-independent manner. TGF-β1 specifically activates TAK1 through the interaction of TβRI with TRAF6, whereas Smad activation is not dependent on TRAF6.
Inactivation of TAK1 by protein phosphatases
It is thought that dysregulated and prolonged TGF-β signaling is implicated in disease states. In order to prevent excessive actions of TGF-β1, a mechanism for efficient downregulation of TAK1 activity would be important. In general, tight regulation of intracellular signaling cascades is accomplished by cyclic phosphorylation and dephosphorylation. In the case of TAK1 inactivation, several members of the Ser/Thr protein phosphatase family have been demonstrated to negatively regulate TAK1 activity. PP2C is capable of binding and dephosphorylating TAK1 in 293 cells under nonstimulated condition [88], [89]. Another Ser/Thr protein phosphatase family member, PP6, interacts with and negatively regulates IL-1-induced TAK1 in 293 cells [90] and TNF-α-induced TAK1 in fibroblasts [91].
We have reported that TAK1 activation by TGF-β1 in glomerular mesangial cells is negatively regulated by another Ser/Thr protein phosphatase family member, PP2A [92], which was previously shown to mediate TGF-β inhibition of p70 S6 kinase (p70S6K) to induce cell-cycle G1 arrest [49]. PP2A associates with TAK1 and TAB1, and Thr-187 in the activation loop of TAK1 is a major dephosphorylation target of PP2A. Our findings in mesangial cells reveal that TAK1 is activated and deactivated very rapidly (Fig. 3). Similar findings have been also reported in cardiac myocytes [93]. Therefore, TAK1 activation is tightly regulated and controlled through rapid phosphorylation and dephosphorylation. In addition, inhibition of PP2A significantly upregulates TAK1 phosphorylation and activity [92], indicating that blockade or attenuation of dephosphorylation by protein phosphatases may cause prolonged activation of TAK1.
TGF-β/TAK1 signaling-mediated fibrotic response
TGF-β1 is believed to be the most potent profibrotic cytokine, and evidence is now accumulating demonstrating that TGF-β1-induced TAK1 signaling plays a critical role in ECM production and the pathogenesis of renal fibrosis. Studies in cultured primary mesangial cells have shown that TGF-β1-induced activation of the MKK3–p38 MAPK cascade leads to type I collagen expression [40], [41] and that TAK1 is a major upstream signaling molecule mediating TGF-β1-induced MKK3 activation and collagen induction [54]. Similarly, TAK1 has been shown to mediate TGF-β-induced expression of types I and IV collagen and fibronectin in cultured immortalized mesangial cells [53]. In fibroblasts, TGF-β-induced fibronectin expression is mediated by TAK1 via the MKK4-JNK signaling cascade [55] and TAK1-deficient fibroblasts exhibit reduced profibrotic response to TGF-β1 stimulation [94]. In addition, p38 MAPK activation is a common mechanism implicated in podocyte injury in proteinuric glomerulopathies induced by puromycin and adriamycin [95]. These studies help to establish TAK1 as a major regulator of TGF-β signaling and pathogenic mechanisms in the renal cellular injury and profibrotic response.
Recent in vivo evidence also provides further support for the critical role of the TAK1 signaling pathway in tissue injury response and fibrosis. Several reports have demonstrated that the MKK3–p38 MAPK and JNK pathways mediate renal inflammation and fibrosis by using pharmacological inhibitors and gene-deficient mice in experimental models of glomerular and tubulointerstitial injury [50], [51], [52], [96], [97], [98], [99], [100], [101]. Increased renal MKK3–p38 MAPK activation has been observed in experimental models of streptozotocin-induced type 1 diabetes and in type 2 diabetic db/db mice, as well as in human diabetic nephropathy [50], [52], whereas Mkk3-deficient db/db mice did not exhibit increased renal p38 MAPK activation and were protected against glomerular injury and fibrosis [50]. These studies suggest that the MKK3–p38 MAPK pathway plays a pathogenic role in diabetic nephropathy. Furthermore, in the unilateral ureteral obstruction (UUO) model of renal fibrosis, blockade of the MKK3–p38 MAPK or JNK pathways resulted in substantial amelioration of and protection against renal inflammation and fibrosis [51], [96], [101]. Inhibition of the p38 MAPK pathway also protected against experimental chronic allograft nephropathy [97], and inhibition of JNK signaling suppressed glomerular and tubulointerstitial damage in the rat antiglomerular basement membrane disease model [98] and in the experimental ischemia/reperfusion model [99], [100].
Examination of human biopsy tissues also provides evidence of increased glomerular p38 MAPK activation in patients with various forms of glomerulonephritis [102], [103], as well as induction of JNK activation following ischemia/reperfusion in human kidney allografts [100] and in various kidney diseases including diabetic nephropathy and IgA nephropathy [99], that paralleled renal injury and implicated p38 MAPK [52], [102], [103] and JNK [99], [100] signaling in the development of inflammation and fibrosis in human kidney disease. Given that TAK1 is a major upstream signaling molecule mediating the activation of the MKK3–p38 MAPK and JNK pathways (see Fig. 2), it stands to reason that TAK1 might play an important role in renal fibrosis in vivo. Indeed, conditional Tak1 gene deletion in mice demonstrates that TAK1 deficiency suppressed interstitial myofibroblast accumulation, collagen deposition, and expression of profibrotic molecules in the UUO kidneys [104]. Collectively, these studies strongly support the notion that TAK1 signaling pathways mediate the development of inflammation, ECM elaboration, and the pathogenesis of renal fibrosis.
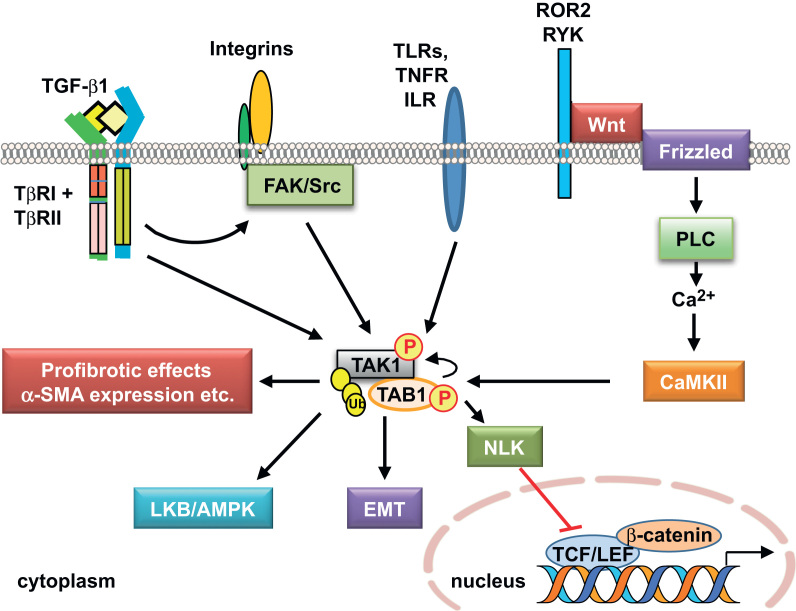
In addition to the kidney, TAK1 signaling is also implicated in profibrotic effects in other organs. TAK1 expressed in the myocardium of transgenic mice enhanced p38 MAPK phosphorylation and promoted interstitial fibrosis and severe myocardial dysfunction [93]. TAK1 has also been identified as a main signaling mediator of epithelial–mesenchymal transition (EMT) and fibrosis in mesothelial epithelial cells from human peritoneum [105], [106]. In addition, TAK1 functions downstream of TGF-β-induced focal adhesion kinase-1 (FAK)/Src activation to mediate fibrotic responses including matrix contraction and expression of α-smooth muscle actin (α-SMA) and profibrotic genes in fibroblasts [94]. By contrast, hepatocyte-specific deletion of the Tak1 gene in mice resulted in spontaneous hepatocyte death, inflammation, fibrosis, and carcinogenesis, indicating that TAK1 signaling can be antifibrotic and is an essential component of cellular homeostasis in the liver [107]. The seemingly opposite effects suggest that TAK1 signaling is, like TGF-β1, capable of exerting dual functions in a tissue/cell type- and context-dependent manner.
Other signaling pathways mediated by TAK1
A summary of additional signaling pathways mediated by TAK1 is depicted in Fig. 4. Recent evidence has shown that TAK1 is activated by agonists of AMP-activated kinase (AMPK) and ischemia, which in turn activates the LKB1/AMPK pathway, a key energy sensor pathway [108]. AMPK is highly expressed in the kidney, where it is thought to be involved in a variety of physiological and pathological processes including ion transport, podocyte function, and diabetic renal hypertrophy [109].
TAK1 has also been shown to negatively regulate the canonical Wnt/β-catenin signaling pathway [110]. Wnt/β-catenin signaling plays an essential role in tissue development, including the kidney, and changes in Wnt ligands and pathway components are associated with acute renal failure and ischemic renal injury, as well as many CKDs, such as diabetic nephropathy, renal fibrosis, kidney cancers, and cystic kidney disease, a class of genetic diseases that includes the most common hereditary life-threatening syndrome polycystic kidney disease [111], [112], [113], [114].
It should be noted that several Wnt proteins are induced simultaneously in response to TGF-β [115], as well as adriamycin [116] or UUO [117], suggesting that Wnt proteins cooperatively function to trigger podocyte injury or interstitial fibrosis, respectively. Wnt-5a activates the TAK1–Nemo-like kinase (NLK) pathway via a noncanonical Wnt/Ca2+ pathway through activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), and antagonizes canonical Wnt/β-catenin signaling [118]. β-Catenin is known to form complexes with members of the T-cell factor/lymphoid enhancer factor (TCF/LEF) classes of transcription factors to regulate the expression of target genes, and the CaMKII–TAK1–NLK pathway inhibits β-catenin-TCF-dependent transcription through phosphorylation of TCF. Although TAK1 is implicated in the negative regulation of canonical Wnt/β-catenin signaling in the Xenopus embryo and in certain mammalian cells including human embryonic kidney HEK293 cells [118], the role of TAK1 in Wnt/β-catenin signaling-mediated renal pathophysiology remains unexplored.
TAK1 signaling in cell fate
TGF-β1 is known to regulate cell survival and cell death, and TAK1 in a similar fashion possesses pro- and antiapoptotic functions. The TAK1-null phenotype is lethal early in embryonic development, and knockdown of TAK1 expression or inhibition of TAK1 activation augments cell apoptosis induced by TGF-β in various cell types in vitro and in vivo, including the kidney, indicating that TAK1 is required for the prevention of apoptosis and plays a role as a cell survival factor [103], [119]. Similarly, TAK1 is essential for preventing the accumulation of TNF-induced reactive oxygen species and the subsequent activation of reactive oxygen species-mediated death pathways in keratinocytes [120]. Conditional tak1 gene deletion in mice resulted in a twofold increase in the apoptotic response in the obstructed kidney after UUO [104]. In contrast, abrogation of TAK1 activation inhibits TGF-β-induced apoptosis in embryonic fibroblasts, prostate cancer cells, and AML12 liver cells, indicating that TAK1 also acts as a promoter of apoptosis [81], [82].
TAK1 signaling in autophagy
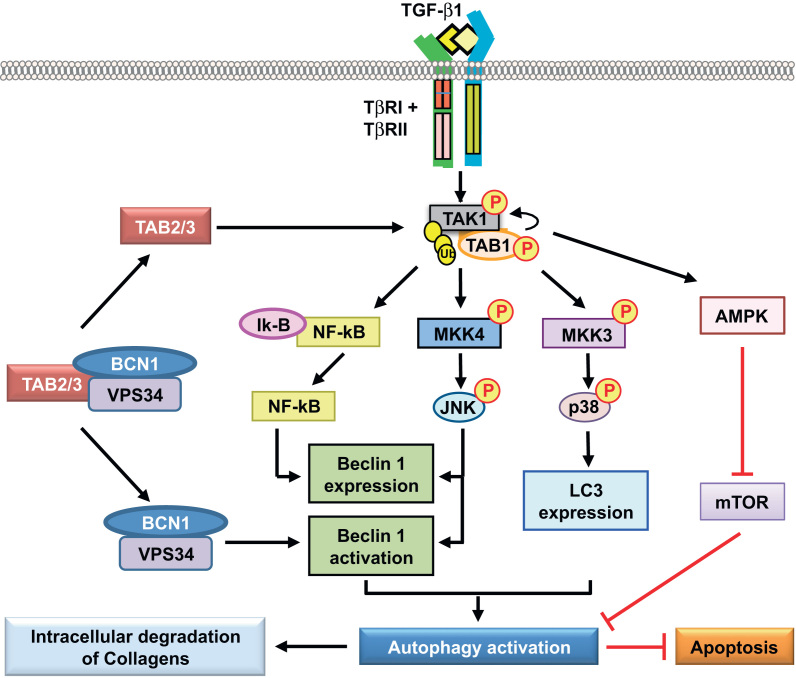
Autophagy, also known as macroautophagy (literally, self-eating), is a fundamental cellular homeostatic process by which cells degrade and recycle proteins and remove damaged organelles [121]. The role of TGF-β1 as an inducer of autophagy is just beginning to be appreciated, and little study has so far been made on the TAK1 signaling pathway in the regulation of autophagy. Autophagy can lead to cell death in response to stress, but it can also act as a protective mechanism for cell survival. It is plausible that the functions of TGF-β1 as both an apoptosis promoter and apoptosis suppressor may relate to its regulation of autophagy. Indeed, it has recently reported that tumor necrosis factor-related apoptosis inducing ligand (TRAIL), which triggers apoptosis preferentially in cancer cells, spares normal untransformed cells from apoptosis by inducing cytoprotective autophagy via the TAK1 signaling pathway [122].
We have reported that TAK1 mediates TGF-β1 induces autophagy, which protects glomerular mesangial cells from undergoing apoptosis during serum deprivation [46]. We also uncovered a novel role of autophagy activated by the TAK1–MKK3–p38 signaling axis as a cytoprotective mechanism to promote intracellular degradation of collagen, and thus negatively regulate and prevent excess collagen accumulation in the kidney [123]. Intriguingly, recent evidence has been demonstrated that two TAK1 binding partners, TAB2 and TAB3, are endogenous inhibitors of autophagy [124], [125]. In nonstimulated conditions, TAB2 and TAB3 are bound with beclin 1, an autophagy-related protein, and maintain the inactive state of beclin 1 and autophagy. In contrast, in response to proautophagic stimuli, TAB2 and TAB3 dissociate from beclin 1 and subsequently bind with TAK1 to facilitate proautophagic stimulation through TAK1 activation (Fig. 5).
Implications for anti-TGF-β therapy
There has been much interest in anti-TGF-β therapy, but although a significant body of evidence in preclinical studies aimed at TGF-β blockade showed great promise of antifibrotic effects in experimental models, limited advances have so far been made in translation to the treatment of human diseases, and the results of most clinical trials have been rather disappointing. A phase I/II randomized, placebo-controlled trial of TGF-β inhibitor therapy using a human anti-TGF-β antibody (Cat-192) in 45 patients with scleroderma failed to show any significant benefits in these patients [126].
Recently, Trachtman et al. reported the results of the first phase I clinical study for the treatment of kidney disease using fresolimumab, a neutralizing anti-TGF-β antibody [127]. Fresolimumab is a human monoclonal antibody that neutralizes all three isoforms of TGF-β. The phase I open-label study was conducted in patients with treatment-resistant primary focal segmental glomerulosclerosis (FSGS) to assess the safety, tolerability, and pharmacokinetics of a single-dose infusion of fresolimumab [127]. The results indicate that fresolimumab is relatively safe and well tolerated in patients, and a larger randomized clinical trial to assess the efficacy of this agent is anticipated.
Pirfenidone is another antifibrotic therapy that is garnering some interest. Its effects are thought to be, in part, mediated by inhibition of TGF-β promoter activity and TGF-β protein secretion, and inhibition of TGF-β-induced Smad2 phosphorylation [128]. In early clinical studies, pirfenidone has shown some promise as a therapy for patients with idiopathic pulmonary fibrosis [129], and to slow the decline of renal function in patients with FSGS [130].
Excessive TGF-β1 activity leads to fibrotic conditions, but, given the complexity and pleiotropic actions of TGF-β1, therapies aimed at indiscriminate blockade of TGF-β1 effects may not be ideal; rather, specific targeting of the TGF-β signaling pathway may arguably be a more effective molecular therapeutic strategy in CKD. Targeting the TAK1 signaling pathway is an attractive strategy that targets the major proinflammatory, proapoptotic, and profibrotic pathways, such as the MMK3–p38 MAPK and JNK, in the treatment of CKD.
In a recently reported preclinical study, the administration of LYTAK1, a selective small-molecule inhibitor of TAK1, plus gemcitabine significantly reduced tumor burden in nude mice with human pancreatic tumor xenografts and prolonged survival duration [131]. Generalizability of the results to human patients is uncertain and too premature at this point. However, it illustrates that targeting of TAK1 is a plausible therapeutic strategy. There is still a great need for future investigations to gain further insights into the complex TGF-β signaling mechanisms, with the ultimate goal of developing novel and more effective therapies for progressive kidney diseases.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






")









