


Introduction
Gait speed (GS) is an important measure of functional ability. Walking is a complex activity that requires integration of motor, sensory, and cognitive functions (CFs). Healthy humans walk at various speeds. However, the ability to regulate speed can diminish with age, neurological deficits, or injuries [1]. Therefore, a slow walking speed is regarded as a strong predictor of adverse outcomes [2]. Assessment of GS is a recommended simple screening tool for functional status in older people, and GS is a major component of sarcopenia or frailty. A slower GS is associated with poor outcomes, including falls, disability, and reduced survival in elderly people [2].
Chronic kidney disease (CKD) is associated with premature aging; and systemic inflammation in patients with CKD is associated with muscle wasting, frailty, cardiovascular disease, and mortality [3]. People with CKD experience comorbidities that are related to changes in gait, such as central nervous system or peripheral neuropathy and cardiopulmonary disease [4]. Therefore, many patients with CKD show numerous gait abnormalities that are influenced by CKD severity [4,5]. However, the factors associated with GS in people with CKD have not been well assessed, and most research is limited to the population on dialysis [4].
The RolE of AST120 (Renamezin) in sarCOpenia preVEntion in pRe-dialYsis CKD patients (RECOVERY) study is a 48-week, randomized controlled, parallel-group, open-label, multicenter trial to determine the role of Renamezin (Daewon Pharmaceutical Co. Ltd.) in sarcopenia-associated factors in patients with CKD (clinicaltrials.gov: NCT03788252). We previously reported that Renamezin had modest effects on GS and quality of life (QoL) [6]. In this study, we investigated the factors associated with GS in patients with CKD with respect to 1) sarcopenic components, 2) plasma uremic and inflammatory markers, and 3) QoL.
Methods
Subjects and design
The RECOVERY protocol has been described elsewhere [6]. The inclusion criteria for RECOVERY participation were: 1) age of ≥20 years; 2) predialysis CKD, serum creatinine of 2.0–5.0 mg/dL, or estimated glomerular filtration rate (eGFR) of 15–60 mL/min/1.73 m2 (calculated based on the Modification of Diet in Renal Disease or Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] equation); 3) serum albumin of ≥3.0 g/dL; 4) no previous history of taking Renamezin in 4 weeks; 5) able to ambulate with or without assistive devices; and 6) willingness to give informed consent. The exclusion criteria were: 1) passage disorders in the gastrointestinal tract and uncontrolled constipation; 2) history of kidney transplantation; 3) use of immunosuppressant agents; 4) uncontrolled hypertension (systolic blood pressure, ≥180 mmHg; diastolic blood pressure, ≥110 mmHg); 5) acute coronary syndrome within three months of the study; 6) acute infectious or inflammatory illness; 7) progressive malignancy; and 8) pregnancy, lactation, or planning to become pregnant during the study period.
A total of 150 study participants were recruited from six general hospitals in the Republic of Korea. Eligible patients who met the inclusion criteria were recruited from Nber 2018 to April 2020. This study was approved by the Institutional Review Board of six medical hospitals and conducted in accordance with the Declaration of Helsinki. All participants provided informed consent. This study was approved by the Institutional Review Board of six medical hospitals, including Chung-Ang University Hospital (No. 1882-003-338), National Medical Center (No. H-1808-093-003), Yeungnam University Hospital (No. YUMC 2018-09-006), CHA Gumi Medical Center (No. GM18-11), Pusan National University Hospital (No. H-1808-003-083), and Dong-A University Hospital (No. DAUHIRB-18-182) and conducted in accordance with the Declaration of Helsinki.
Measurement and definitions
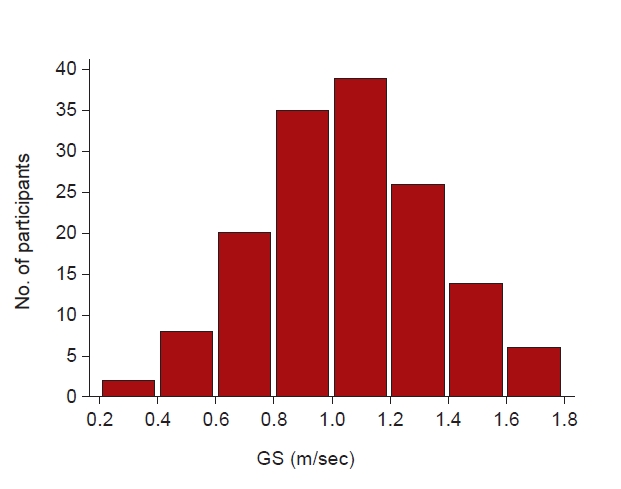
GS was measured over 6-m with a dynamic start. Participants walked at a normal walking speed under the instructions of the examiner. Participants started walking 2 m before the measurement point (acceleration zone) and ended 2 m after the measurement point (deceleration zone). Participants repeated each type of walking speed test twice, and the mean data were analyzed. GS was classified into four groups; cutoffs for GS were ≤0.8, 0.8–1.0, ≤1.0–1.3, and ≥1.3 m/sec. The cutoff speed of ≤0.8 m/sec was based on the European Working Group on Sarcopenia criteria [7], <1.0 m/sec was based on the Asian Working Group on Sarcopenia criteria [8], and >1.3 m/sec was established for extremely fit individuals [9]. Handgrip strength (HGS) was assessed using a digital handgrip dynamometer (T.K.K.5401; Takei Scientific Instruments Co., Ltd.) in both hands. Participants measured HGS while standing, and participants were encouraged to grasp the digital handgrip dynamometer strongly three times at intervals of 30 seconds each way. The greatest HGS values were used in the analysis. Body composition was measured using a bioelectrical impedance analysis device (InBody S10; InBody Co., Ltd.) [10]. Skeletal muscle mass index (SMI) was calculated by dividing the limb skeletal muscle mass (kg) by the square of the height (m2). For QoL assessment, the Kidney Disease Quality of Life Short Form (KDQOL-SF), 1.3 Korean version [11] was used. Briefly, this assessment consists of kidney disease (KD)-targeted scales and SF-36. KD-targeted scales include symptoms/problems (Sx), effects of KD, burden of KD, work status (WS), CF, quality of social interaction (QSI), sexual function, sleep, social support (SS), dialysis staff encouragement, and patient satisfaction. The SF-36 includes eight multi-item scales that summarize a physical component scale (PCS) and a mental component scale (MCS). The PCS encompasses physical functioning (PF), role limitations caused by physical health problems (RP), bodily pain (BP), and general health. The MCS encompasses vitality (energy/fatigue), social functioning (SF), role limitations due to emotional problems (RE), and mental health (psychological distress and psychological well-being). Physical activity was assessed using the International Physical Activity Questionnaire.
The levels of parameters, including serum indoxyl sulfate (IS), tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), myostatin, intact parathyroid hormone (iPTH), and 25-OH-vitamin D, were measured at the central laboratory institution (Seoul Clinical Laboratories in Yongin, Republic of Korea). Myostatin levels were assessed by enzyme-linked immunosorbent assay (ELISA) using DGDF80 (GDF-8/Myostatin Quantikine ELISA Kit; R&D Systems), IL-6 levels by ELISA using HS600C (Human IL-6 Quantikine HS ELISA Kit; R&D Systems), and TNF-α levels by ELISA using HSTA00D (Human TNF-α Quantikine HS ELISA; R&D Systems). The iPTH level was measured using an electrochemiluminescence immunoassay, and 25-OH vitamin D levels were measured by a chemiluminescence immunoassay using a Cobas E801 analyzer (Roche Diagnostics GmbH). Serum IS levels were measured using a high-performance liquid chromatography-fluorescence detector (HPLC-FLD, Agilent 1100 series; Agilent Technologies). Other blood and urine data were collected from each research institution. The eGFR was calculated using the CKD-EPI equation [12].
Statistical analyses
Means ± standard deviations were calculated for continuous variables, and the differences were determined by the Kruskal-Wallis test. The skewed variables were normally distributed after natural logarithm transformation. The chi-square test was used for categorical variables. The Jonckheere-Terpstra test and linear by linear association were used to calculate the p-value to determine the trends for continuous and categorical variables, respectively. Statistical significance was set at p < 0.05. Logistic regression analysis was used to identify factors associated with GS. The levels of inflammatory and uremic markers are expressed as box-and-whisker plots. Statistical analyses were performed using IBM SPSS Statistics version 22.0 (IBM Corp.).
Results
Clinical characteristics of the study participants
The mean GS was 1.06 ± 0.3 m/sec. Fig. 1 shows the distribution of GS for all participants. The mean age of the total population was 65.0 ± 10.8 years, and 97 participants (64.7%) were male. Seventy-seven participants (51.3%) had diabetes mellitus (DM), and 22 (14.7%) were in the “medical aid” insurance status group. The mean serum creatinine level was 2.09 ± 0.73 mg/dL, and the eGFR was 33.8 ± 12.5 mL/min/1.73 m2.
The baseline demographics and laboratory parameters of the patients grouped according to GS are shown in Table 1. The group with a GS of ≤0.8 m/sec was the oldest, had the highest proportion of participants with low education level, and had the highest proportion receiving medical aid. The level of hemoglobin was the lowest and that of IL-6 was the highest in the group with a GS of ≤0.8 m/sec. The levels of albumin, creatinine, eGFR, proteinuria, and metabolic equivalent of task scale did not differ among the groups. Further subgroup analysis by sex and by the cutoff value of 1.0 m/sec are shown in Supplementary Table 1–3 (available online).
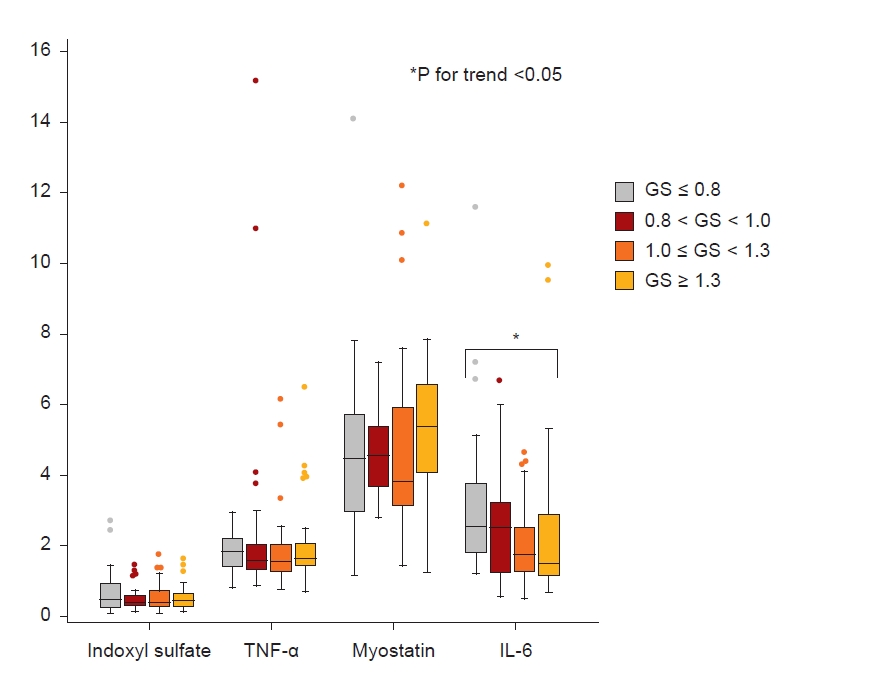
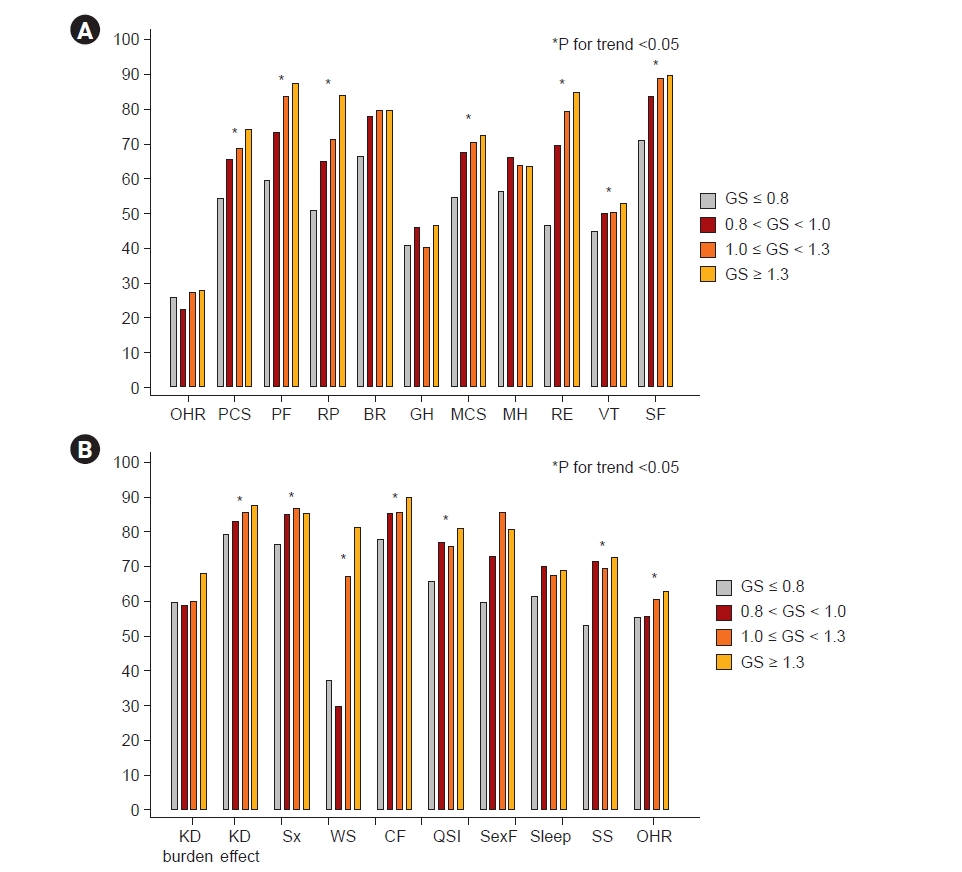
Fig. 2 shows the levels of IS, TNF-α, myostatin, and IL-6 among the four groups. The levels of IS, TNF-α, and myostatin were not different among the groups; however, the level of IL-6 trended higher in the lowest GS group. In the KDOQL-SF, participants with a GS of ≤0.8 m/sec had the lowest PCS and MCS scores (Table 1, Fig. 3). Specifically, the items with a positive trend according to GS were: the level of PF, RP in PCS, RE, vitality (VT), SF in MCS, KD effect, Sx, WS, CF, QSI, SS, and overall health rating (OHR) on a KD-specific scale (Fig. 3; Supplementary Table 4, available online).
Associated factors with gait speeds
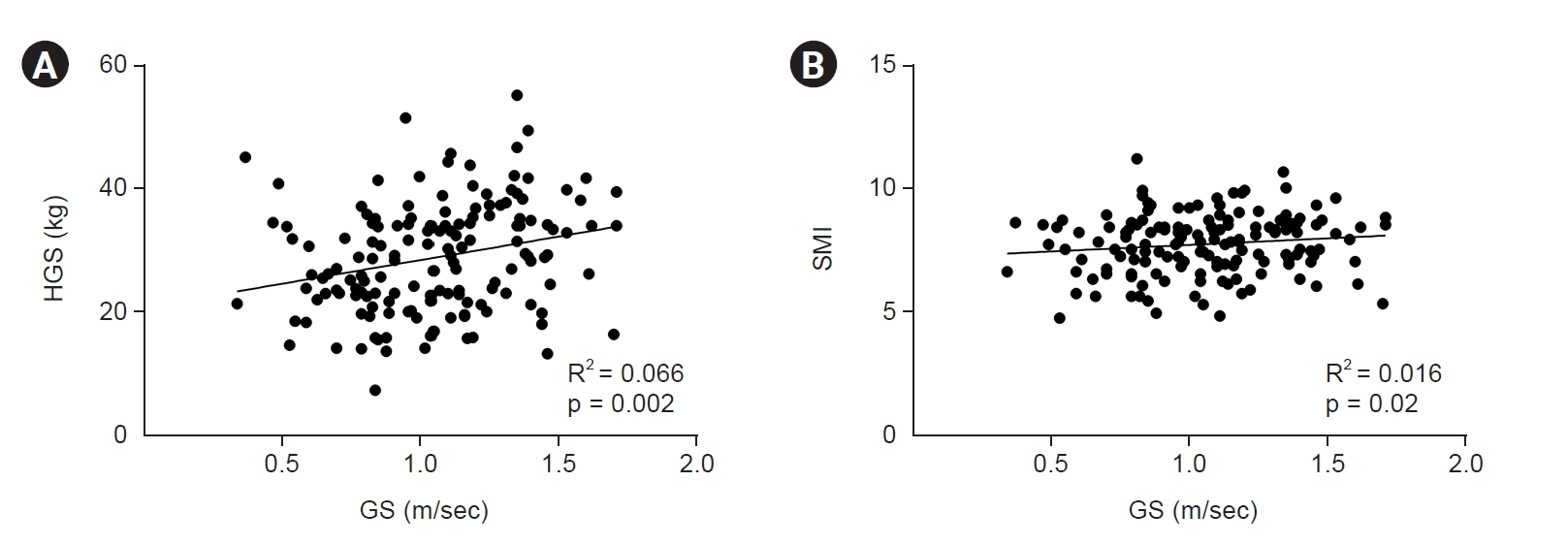
We examined the relationship between the sarcopenia factors. We performed a correlation analysis to determine the relationship between GS and HGS or the SMI (Fig. 4). HGS and GS showed a weak correlation (R2 = 0.066, p < 0.001); however, GS and the SMI were not significantly associated (R2 = 0.016, p = 0.119). In the linear regression analysis adjusted for age, sex, history of DM, and eGFR, HGS was significantly associated with GS (β = 0.85 [95% CI, 0.08–1.63], p = 0.03); however, the SMI showed no association with GS (β =1.87 [95% CI, –2.92 to 6.66], p = 0.44) (Table 2).
Next, we evaluated the relationship between GS and KDQOL-SF. OHR was not significantly associated with GS. However, some PCS (β = 0.51 [95% CI, 0.27–0.74], p < 0.001) and MCS (β = 0.45 [95% CI, 0.21–0.69], p < 0.001) factors were significantly associated with GS in the multivariate analysis adjusted for age, sex, DM, and eGFR. Specifically, PF, RP, and BP in the PCS and RE, VT, and SF in the MCS were statistically significantly correlated with GS. In the KD-specific scale, Sx, WS, CF, QSI, SS, and OHR were statistically significantly correlated with GS in the multivariate analysis (Supplementary Table 5, available online).
As for laboratory findings, the levels of hemoglobin (β = 2.84 [95% CI, 0.12–5.56], p = 0.04) and IL-6 (β = –13.2 [95% CI, –21.1 to –5.25], p = 0.001) were significantly associated with GS in multivariate analysis adjusted for age, sex, DM, and eGFR. In contrast, the levels of TNF-α, myostatin, and IS were not statistically significantly associated with GS (Table 2). Further analysis according to sex was described in Supplementary Table 6 (available online).
Discussion
Walking speed is a simple and sensitive indicator of overall functional capacity. In this study, we identified several factors associated with GS in 150 patients with predialysis CKD. We specifically focused on the association between GS and sarcopenia components; uremic and inflammatory biomarkers, such as IS, TNF-α, myostatin, and IL-6; and QoL. GS had a weak statistically significant association with HGS, but not with SMI. IL-6 was statistically significantly associated with GS but not IS, TNF-α, or myostatin. In the KDQOL, both PCS and MCS were statistically significantly associated with GS. This study demonstrated that a variety of factors are related to GS in predialysis patients with CKD.
GS is regarded as a functional vital sign. GS is a valid and reliable measure appropriate for assessing and monitoring functional status and overall health. GS has various cutoff values that are indicative of specific outcomes, such as cognitive decline, hospitalization, or functional dependence [2]. Thus, the primary outcome of the RECOVERY study was the GS difference of ≥0.1 m/sec between the control group and the Renamezin group [6]. This study analyzed baseline GS when the patients were registered.
In this study, GS was statistically significantly correlated with HGS, but not with the SMI. Sarcopenia is defined as low muscle function and low muscle mass. The impact of muscle mass on clinical outcomes has been well described in previous studies [13,14]; however, muscle function is more important than muscle mass in terms of patient outcomes, and loss of muscle mass alone may not fully reflect muscular function [15]. Another study reported that muscle mass was not associated with GS [16,17]. Muscle mass and muscle quality are difficult to measure accurately. Body impedance analysis has limitations that include suboptimal diagnostic accuracy [7,13].
Sarcopenia is associated with increased inflammatory activity, including that of proinflammatory and anti-inflammatory cytokines. We assessed several uremic and inflammatory biomarkers and found that IL-6 was associated with GS. IL-6 is a circulating cytokine secreted from several different cells including activated macrophages, lymphocytes, and adipose tissue. IL-6 plays a major role in the differentiation of CD4-positive T cell subsets and production of acute phase proteins and acts as an essential factor in bone homeostasis and metabolic control [18]. The IL-6 pathway is one of the main signaling pathways that modulate the relationship between aging and chronic morbidity [19]. In experimental studies, elevated IL-6 levels in the plasma led to lipolysis in skeletal muscle and systemic fatty acid oxidation [20]. Additionally, many clinical studies have supported the association of IL-6 with sarcopenia or GS. A study on 333 adults aged ≥70 years showed that higher IL-6 levels were associated with slower gait velocity, and the highest IL-6 quartile showed a faster decline in GS [21]. In the Health, Aging and Body Composition study, IL-6 levels predicted a functional decline [19]. A reduction in IL-6 levels was associated with GS improvement [22].
TNF-α was not associated with GS in this study. TNF-α has been reported to be associated with a decline in both muscle mass and muscle strength [23]. However, TNF-α is a less stable biomarker, as suggested by the high inter-assay coefficient of variation [24]. Moreover, the tissue expression of TNF-α may not be matched by increased serum levels. Myostatin, a potential biomarker of muscle wasting for sarcopenia and cachexia, showed conflicting results regarding its association with body composition, muscle mass, and strength [25]. A previous study also reported no association between myostatin levels and GS [26]. The abundance of myostatin does not exhibit any clear activity; and the level of myostatin depends on many factors, such as growth of skeletal muscle mass, aging, weight loss, or kidney function [27]. IS, a uremic toxin derived from the metabolism of tryptophan, has been examined for its potential role in uremic sarcopenia in experimental studies [28,29]. However, clinical studies have failed to demonstrate a consistent relationship between sarcopenia and IS [28,30]. The level of IS showed no difference in the GS group of this study. The authors speculate that this is a product of there being no difference in eGFR between groups. IS is a representative uremic toxin, the level of which depends on eGFR. We could not reach a clear conclusion when considering serum biomarkers and GS due to the small sample size, and further studies are needed.
Both PCS and MCS were associated with low GS in patients with CKD. This is in line with a previous study that showed that GS correlated not only with physical function but also mood or cognition [31]. Several components of the KDQOL were correlated with GS in this study. Specifically, social (SF, SS, and QSI) or psychological (RE and VT) problems could affect GS. Recently, the multidomain approach to frailty has received attention. Physical frailty and psychological or social frailty are interactive and cause poor outcomes, such as mortality, hospitalization, or disability [32]. GS could provide clues to various health problems experienced.
Interestingly, GS was associated with low socioeconomic status (SES), such as receipt of medical aid or low education level, which is consistent with previous studies. A multi-cohort population-based study reported that a 60-year-old man with low SES had the same walking speed as a 66.6-year-old man with a high SES [33]. In women’s health and aging studies, older women with less than a high school degree had threefold greater odds of frailty than more educated individuals [34]. Socioeconomic inequalities could be an important barrier to healthy aging. People who have experienced socioeconomic adversity are not only more likely to live shorter lives but also to live more of their shorter lives with disability [35]. There are several biological mechanisms that could explain the relationship between SES and frailty. SES has been linked to inflammation [36] and may be linked to frailty through decreased physical activity or poor nutrition [37–39]. We cannot improve socioeconomic circumstances for CKD patients, but we can recognize that SES can be a significant issue.
Our study has some limitations. This was a cross-sectional study. We were unable to determine the causal effect of the relationship between GS and outcomes. The study population was too small to result in definitive conclusions. The eGFR was calculated based on serum creatinine level, which was affected by muscle mass. Furthermore, muscle mass assessed by impedance analysis has suboptimal diagnostic accuracy. However, our current findings do reveal various markers associated with GS in patients with predialysis CKD.
In conclusion, GS was associated with an inflammatory marker, IL-6, and with QoL measures, MCS and PCS, in predialysis CKD patients. Further studies with a longer follow-up are required.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement table 1
Supplement table 1 Print
Print






")









